Les 5
Leeruitkomsten
- De student kan comma-separated values (CSV) bestanden inlezen in R.
- De student kan bepalen welke categoriën een categorische variabele bevat en hoeveel waarnemingen per categorie er zijn met de
distinctencountfuncties. - De student kan twee datasets samenvoegen met de
left_joinfunctie. - De student kan de volgorde van categoriën op de x-as in een boxplot of staafdiagram aanpassen.
Voorbereiding
Opdracht 5
Maak een nieuw R project aan voor les 5.
Data inlezen
Naast TSV en Excel bestanden is er nog een bestandstype dat veel gebruikt wordt voor de opslag van data. Dit is de comma-separated value (CSV) bestand. Er zijn twee soorten CSV files:
- In de meeste Engelstalige landen is een CSV bestand een bestand waar de
,als scheidingsteken wordt gebruikt voor het scheiden van de kolommen. Dit soort bestanden kun je inlezen met de functieread_csvuit hettidyversepackage. - In enkele Europese landen (waaronder Nederland) wordt de
,gebruikt als decimaalscheidingsteken. In deze landen is een CSV bestand een bestand waar de;als scheidingsteken wordt gebruikt voor het scheiden van de kolommen. Deze bestanden kun je inlezen met de functieread_csv2uit hettidyversepackage.
Je kunt CSV bestanden herkennen aan de file extensie .csv, maar deze file extensie is niet altijd aanwezig. Bovendien zijn er dus twee soorten CSV bestanden. Zorg er dus voor dat je altijd controleert wat het gebruikte scheidingsteken is door het bestand in RStudio te openen (zie les 2).
Laten we een voorbeeld bekijken. De dataset toothgrowth.csv bevat informatie over tandgroei in cavia’s. De length_um variabele geeft de lengte van de odontoblasten in micrometer. Odontoblasten zijn de cellen die ervoor zorgen dat tanden groeien. Verder geven de supplement en de dose_mgperday variabele aan welke behandeling het dier kreeg; de dieren kregen vitamine C toegediend in pure vorm (supp is VC) of in de vorm van sinaasappelsap (supp is OJ) en in verschillende doses (dose is 0.5, 1 of 2 mg/dag).
We gaan deze dataset inlezen in R. De file extensie doet vermoeden dat het een CSV bestand is. Als we het bestand openen in RStudio zien we dat de komma (,) is gebruikt als kolomscheidingsteken en de punt (.) als decimaalscheidingsteken. We gebruiken daarom de read_csv functie om het bestand in te lezen:
## Rows: 60 Columns: 3
## ── Column specification ────────────────────────────────────────────────────────────────────────────────
## Delimiter: ","
## chr (1): supplement
## dbl (2): length_um, dose_mgperday
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.Net als bij de andere read_ functies uit tidyverse wordt de data ingelezen als tibble:
## # A tibble: 60 × 3
## length_um supplement dose_mgperday
## <dbl> <chr> <dbl>
## 1 4.2 VC 0.5
## 2 11.5 VC 0.5
## 3 7.3 VC 0.5
## 4 5.8 VC 0.5
## 5 6.4 VC 0.5
## 6 10 VC 0.5
## 7 11.2 VC 0.5
## 8 11.2 VC 0.5
## 9 5.2 VC 0.5
## 10 7 VC 0.5
## # ℹ 50 more rowsOpdracht 5
Het bestand salary.csv bevat data over het salaris van verschillende medewerkers van een bedrijf. Lees dit bestand in in R.
Klik hier voor het antwoord
De file extensie doet vermoeden dat het een CSV bestand is. Als we het bestand openen in RStudio zien we dat de komma (;) is gebruikt als kolomscheidingsteken en de punt (,) als decimaalscheidingsteken. We gebruiken daarom de read_csv2 functie om het bestand in te lezen:
## ℹ Using "','" as decimal and "'.'" as grouping mark. Use `read_delim()` for more control.## Rows: 10 Columns: 4
## ── Column specification ────────────────────────────────────────────────────────────────────────────────
## Delimiter: ";"
## dbl (3): employee, group, salaryincrease_percentage
## num (1): salary_dollar
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.Data inspecteren
Bepalen welke categoriën er zijn met distinct
Voor categorische variabelen in een dataset is het nuttig om tijdens de data inspectie te bepalen welke categoriën de variabele bevat. We kunnen hiervoor de distinct functie uit het tidyverse package gebruiken. Om bijvoorbeeld de verschillende supplementen in de tandgroei dataset te bekijken, kunnen we het volgende doen:
## # A tibble: 2 × 1
## supplement
## <chr>
## 1 VC
## 2 OJBepalen hoeveel waarnemingen per categorie er zijn met count
Vaak wil je niet alleen weten welke categoriën er zijn, maar wil je ook weten hoeveel observaties elke categorie in de data bevat. Hiervoor kunnen we de count functie uit het tidyverse package gebruiken. Om te zien hoeveel observaties er per supplement in de tandgroei data zijn doen we het volgende:
## # A tibble: 2 × 2
## supplement n
## <chr> <int>
## 1 OJ 30
## 2 VC 30We kunnen het aantal observaties voor de supplementen ook opsplitsen voor de dosis:
## # A tibble: 6 × 3
## supplement dose_mgperday n
## <chr> <dbl> <int>
## 1 OJ 0.5 10
## 2 OJ 1 10
## 3 OJ 2 10
## 4 VC 0.5 10
## 5 VC 1 10
## 6 VC 2 10Scenario 3 voor niet-tidy data: meerdere variabelen in één cel
In les 3 hebben we twee scenario’s gezien voor niet-tidy data en hebben we gebruik gemaakt van de functies pivot_wider en pivot_longer om de data tidy te maken. Er is nog een derde scenario: datasets waarin meerdere variabelen in één cel staan. Een voorbeeld is de table3 dataset die standaard wordt geladen als je het tidyverse package laadt:
## # A tibble: 6 × 3
## country year rate
## <chr> <dbl> <chr>
## 1 Afghanistan 1999 745/19987071
## 2 Afghanistan 2000 2666/20595360
## 3 Brazil 1999 37737/172006362
## 4 Brazil 2000 80488/174504898
## 5 China 1999 212258/1272915272
## 6 China 2000 213766/1280428583In deze dataset bevatten de cellen van de laatste kolom meerdere variabelen, namelijk het aantal tuberculosis gevallen en het totale aantal inwoners.
Om deze vorm van niet-tidy data om te zetten naar tidy data kunnen we gebruik maken van de separate functie. Deze functie splits een kolom in twee kolommen op basis van een scheidingsteken:
table3_tidy <- table3 |>
separate(col = rate, into = c("tuberculosiscases", "population"),
sep = "/", convert = TRUE)
table3_tidy## # A tibble: 6 × 4
## country year tuberculosiscases population
## <chr> <dbl> <int> <int>
## 1 Afghanistan 1999 745 19987071
## 2 Afghanistan 2000 2666 20595360
## 3 Brazil 1999 37737 172006362
## 4 Brazil 2000 80488 174504898
## 5 China 1999 212258 1272915272
## 6 China 2000 213766 1280428583We geven in de separate functie aan welke kolom opgesplitst moet worden (col argument) en wat de kolomnamen zijn van de nieuwe kolommen (into argument). Verder geven we op basis van welk scheidingsteken de kolom opgesplitst moet worden (sep argument) en dat getallen als getal, en niet als tekst, moeten worden weergegeven (convert = TRUE).
Opdracht 5
Het bestand iris.csv bevat data over de bloembladlengtes (in centimeter) van verschillende soorten irissen (we zijn deze data ook al tegen gekomen in de casus van les 1). Voer voor dit bestand de volgende stappen uit:
- Lees de data in in R.
- Bepaal of de dataset tidy is. Zo niet, maak de data dan tidy.
- Bepaal hoeveel waarnemingen er voor elke soort zijn.
Klik hier voor het antwoord
De file extensie doet vermoeden dat het een CSV bestand is. Als we het bestand openen in RStudio zien we dat de komma (,) is gebruikt als kolomscheidingsteken en de punt (.) als decimaalscheidingsteken. We gebruiken daarom de read_csv functie om het bestand in te lezen:
## Rows: 150 Columns: 3
## ── Column specification ────────────────────────────────────────────────────────────────────────────────
## Delimiter: ","
## chr (3): sepallengthandwidth_cm, petallengthandwidth_cm, species
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.De dataset is niet tidy. De kolommen met data over de sepals en petals bevatten namelijk meerdere variabelen per cel (scenario 3 voor niet-tidy data). We moeten deze kolommen dus opsplitsen:
# Splits de kolom met sepal meetwaarden in twee aparte kolommen
iris <- iris |>
separate(col = sepallengthandwidth_cm, into = c("sepallength_cm", "sepalwidth_cm"),
sep = "/", convert = TRUE)
# Splits de kolom met petal meetwaarden in twee aparte kolommen
iris <- iris |>
separate(col = petallengthandwidth_cm, into = c("petallength_cm", "petalwidth_cm"),
sep = "/", convert = TRUE)
# Bekijk de data
iris## # A tibble: 150 × 5
## sepallength_cm sepalwidth_cm petallength_cm petalwidth_cm species
## <dbl> <dbl> <dbl> <dbl> <chr>
## 1 5.1 3.5 1.4 0.2 setosa
## 2 4.9 3 1.4 0.2 setosa
## 3 4.7 3.2 1.3 0.2 setosa
## 4 4.6 3.1 1.5 0.2 setosa
## 5 5 3.6 1.4 0.2 setosa
## 6 5.4 3.9 1.7 0.4 setosa
## 7 4.6 3.4 1.4 0.3 setosa
## 8 5 3.4 1.5 0.2 setosa
## 9 4.4 2.9 1.4 0.2 setosa
## 10 4.9 3.1 1.5 0.1 setosa
## # ℹ 140 more rowsNu de data tidy is, kunnen we bepalen hoeveel observaties er zijn per soort:
## # A tibble: 3 × 2
## species n
## <chr> <int>
## 1 setosa 50
## 2 versicolor 50
## 3 virginica 50Voor elke soort zijn dus 50 observaties beschikbaar.
Data bewerken
Twee datasets samenvoegen met left_join
In sommige gevallen is de data opgesplitst in twee delen. We kunnen dan beide delen samenvoegen door gebruik te maken van de left_join functie. Laten we een voorbeeld bekijken om te zien hoe dit werkt. We kijken hiervoor naar een aangepaste versie van de tandgroei data:
## Rows: 60 Columns: 4
## ── Column specification ────────────────────────────────────────────────────────────────────────────────
## Delimiter: ","
## chr (2): identifier, supplement
## dbl (2): length_um, dose_mgperday
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.## # A tibble: 60 × 4
## identifier length_um supplement dose_mgperday
## <chr> <dbl> <chr> <dbl>
## 1 guineapig001 4.2 VC 0.5
## 2 guineapig002 11.5 VC 0.5
## 3 guineapig003 7.3 VC 0.5
## 4 guineapig004 5.8 VC 0.5
## 5 guineapig005 6.4 VC 0.5
## 6 guineapig006 10 VC 0.5
## 7 guineapig007 11.2 VC 0.5
## 8 guineapig008 11.2 VC 0.5
## 9 guineapig009 5.2 VC 0.5
## 10 guineapig010 7 VC 0.5
## # ℹ 50 more rowsIn principe is dit dezelfde data als de data die we eerder hebben bekeken. We zien alleen een extra kolom met een identifier, een combinatie van letters en cijfers die voor elke rij uniek is. Er is ook nog een andere dataset, met daarin informatie over de vachtkleur van de dieren:
## Rows: 60 Columns: 2
## ── Column specification ────────────────────────────────────────────────────────────────────────────────
## Delimiter: ","
## chr (2): identifier, furcolour
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.## # A tibble: 60 × 2
## identifier furcolour
## <chr> <chr>
## 1 guineapig001 black
## 2 guineapig002 black
## 3 guineapig003 white
## 4 guineapig004 black
## 5 guineapig005 brown
## 6 guineapig006 black
## 7 guineapig007 brown
## 8 guineapig008 white
## 9 guineapig009 brown
## 10 guineapig010 black
## # ℹ 50 more rowsWe willen deze twee datasets nu samenvoegen tot één dataset. We gebruiken hiervoor de left_join functie:
tandgroei_compleet <- left_join(tandgroei_deel1, tandgroei_deel2, by = "identifier")
tandgroei_compleet## # A tibble: 60 × 5
## identifier length_um supplement dose_mgperday furcolour
## <chr> <dbl> <chr> <dbl> <chr>
## 1 guineapig001 4.2 VC 0.5 black
## 2 guineapig002 11.5 VC 0.5 black
## 3 guineapig003 7.3 VC 0.5 white
## 4 guineapig004 5.8 VC 0.5 black
## 5 guineapig005 6.4 VC 0.5 brown
## 6 guineapig006 10 VC 0.5 black
## 7 guineapig007 11.2 VC 0.5 brown
## 8 guineapig008 11.2 VC 0.5 white
## 9 guineapig009 5.2 VC 0.5 brown
## 10 guineapig010 7 VC 0.5 black
## # ℹ 50 more rowsBeide datasets zijn nu samengevoegd tot één dataset. De left_join functie heeft hiervoor gebruik gemaakt van de identifier kolommen in beide datasets. Doordat de waarden in deze kolommen in beide datasets overeenkomen, kan de left_join functie met deze kolommen bepalen welke rijen in beide datasets bij elkaar horen. Het is daarom heel belangrijk dat hier een kolom wordt gebruikt waarvan de waarden uniek zijn voor elke rij.
Opdracht 5
Een onderzoeker voert een fluorescentieassay uit in een 96 wellsplaat. Hierin wordt het effect van acht verschillende chemicaliën in vier verschillende concentraties onderzocht. Alle metingen zijn in triplo uitgevoerd. De data is te vinden in het bestand screening.xlsx. Een sheet in het Excel bestand bevat de experimentele opzet. De andere sheet bevat de meetresultaten.
Voer de volgende stappen uit:
- Lees beide sheets in in R.
- Combineer beide datasets in één tibble.
Klik hier voor het antwoord
De data staat in een Excel bestand. We lezen de data daarom in met de read_excel functie:
screening_opzet <- read_excel("screening.xlsx", sheet = "experiment")
screening_metingen <- read_excel("screening.xlsx", sheet = "data")Om beide datasets samen te voegen moeten we gebruik maken van een kolom met unieke waarden.
## # A tibble: 96 × 3
## well_id chemical concentration
## <chr> <chr> <dbl>
## 1 a1 zzz1 0
## 2 a2 zzz1 0
## 3 a3 zzz1 0
## 4 a4 zzz1 10
## 5 a5 zzz1 10
## 6 a6 zzz1 10
## 7 a7 zzz1 50
## 8 a8 zzz1 50
## 9 a9 zzz1 50
## 10 a10 zzz1 100
## # ℹ 86 more rows## # A tibble: 96 × 2
## well_id fluorescentie
## <chr> <dbl>
## 1 a1 111
## 2 a2 150
## 3 a3 136
## 4 a4 112
## 5 a5 145
## 6 a6 147
## 7 a7 155
## 8 a8 139
## 9 a9 146
## 10 a10 186
## # ℹ 86 more rowsIn dit geval is dat in beide datasets de kolom well_id. We gebruiken de left_join functie om beide datasets samen te voegen:
screening_compleet <- left_join(screening_opzet, screening_metingen, by = "well_id")
screening_compleet## # A tibble: 96 × 4
## well_id chemical concentration fluorescentie
## <chr> <chr> <dbl> <dbl>
## 1 a1 zzz1 0 111
## 2 a2 zzz1 0 150
## 3 a3 zzz1 0 136
## 4 a4 zzz1 10 112
## 5 a5 zzz1 10 145
## 6 a6 zzz1 10 147
## 7 a7 zzz1 50 155
## 8 a8 zzz1 50 139
## 9 a9 zzz1 50 146
## 10 a10 zzz1 100 186
## # ℹ 86 more rowsResultaten communiceren
Volgorde van de categoriën op de x-as aanpassen
Als we met ggplot2 een staafdiagram maken, wordt de volgorde van de categoriën op de x-as bepaald door de namen van de categoriën (alfabetische volgorde). Laten we als voorbeeld een staafdiagram maken voor het aantal dieren per vachtkleur in de tandgroeidataset:
tandgroei_compleet |> count(furcolour) |>
ggplot(aes(x = furcolour, y = n)) +
geom_col() +
theme_minimal() +
labs(
title = "Vachtkleur van de cavia's in de tandgroei dataset",
x = "Vachtkleur",
y = "Aantal dieren"
)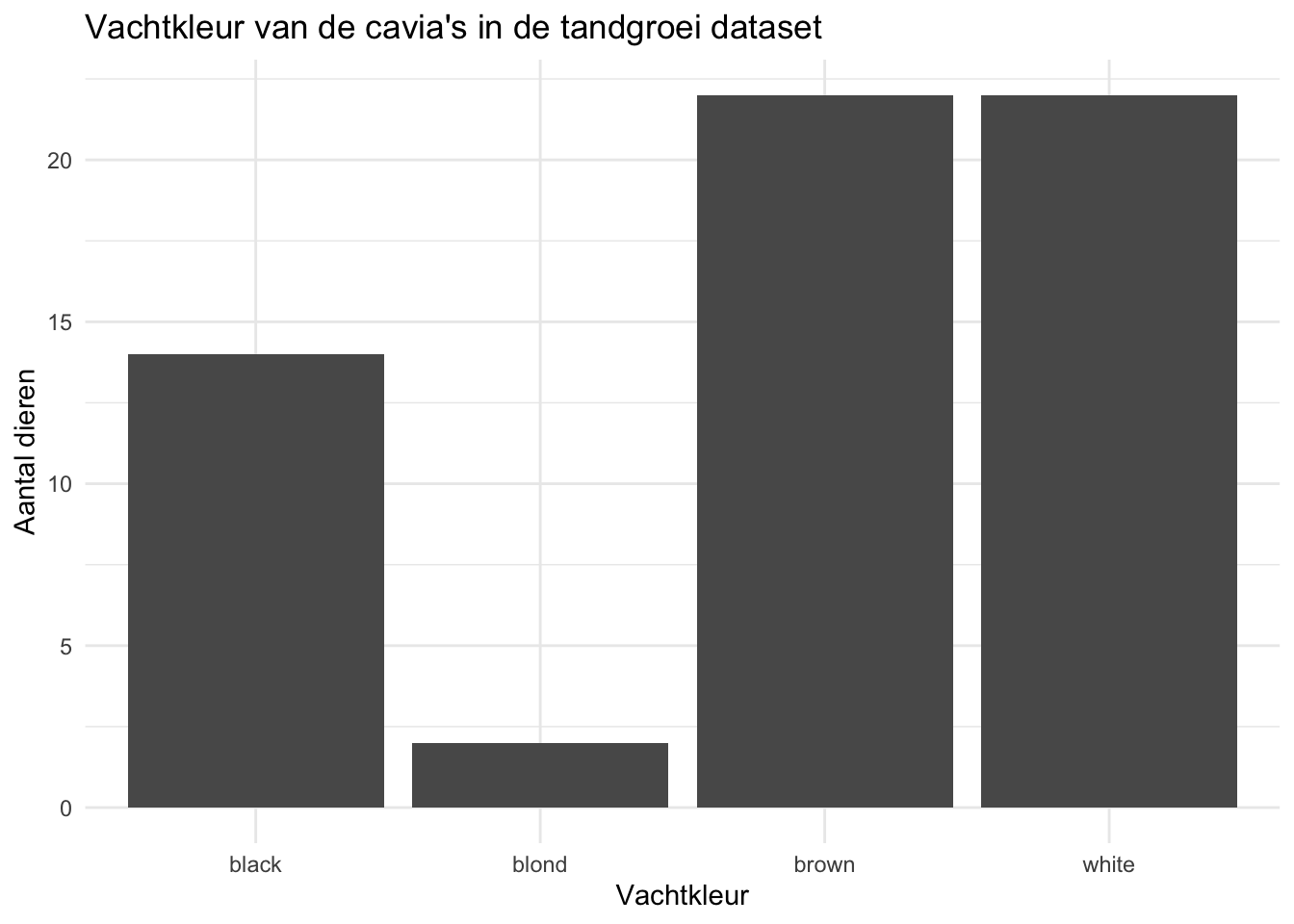
Zoals te zien is worden de categoriën op de x-as op alfabetische volgorde gezet. We kunnen deze volgorde aanpassen. We kunnen bijvoorbeeld de categoriën ordenen op basis van het aantal dieren in elke categorie met de reorder functie. We kunnen ze bijvoorbeeld sorteren van klein naar groot:
tandgroei_compleet |> count(furcolour) |>
ggplot(aes(x = reorder(furcolour, n), y = n)) +
geom_col() +
theme_minimal() +
labs(
title = "Vachtkleur van de cavia's in de tandgroei dataset",
x = "Vachtkleur",
y = "Aantal dieren"
)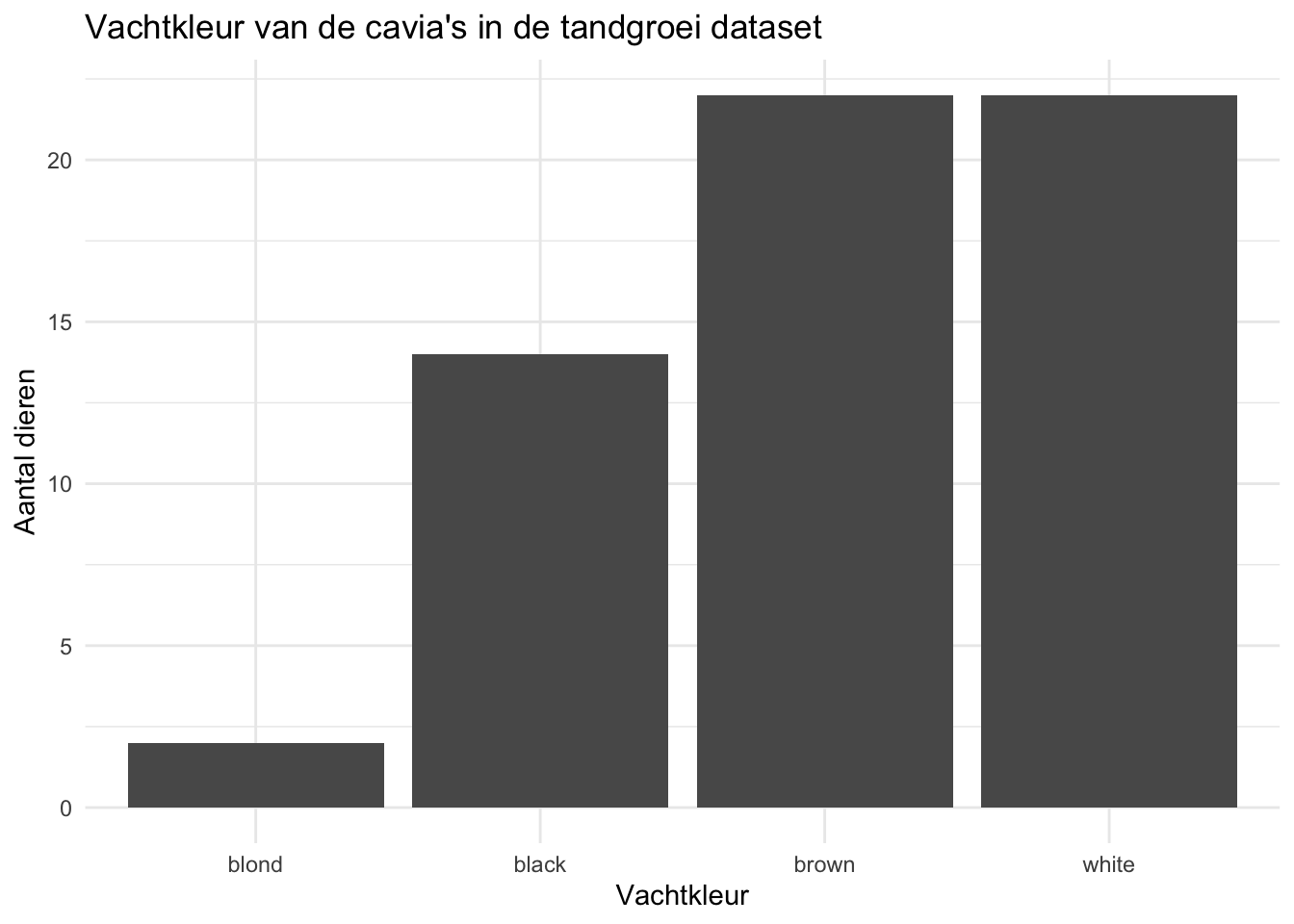
Of juist van klein naar groot:
tandgroei_compleet |> count(furcolour) |>
ggplot(aes(x = reorder(furcolour, -n), y = n)) +
geom_col() +
theme_minimal() +
labs(
title = "Vachtkleur van de cavia's in de tandgroei dataset",
x = "Vachtkleur",
y = "Aantal dieren"
)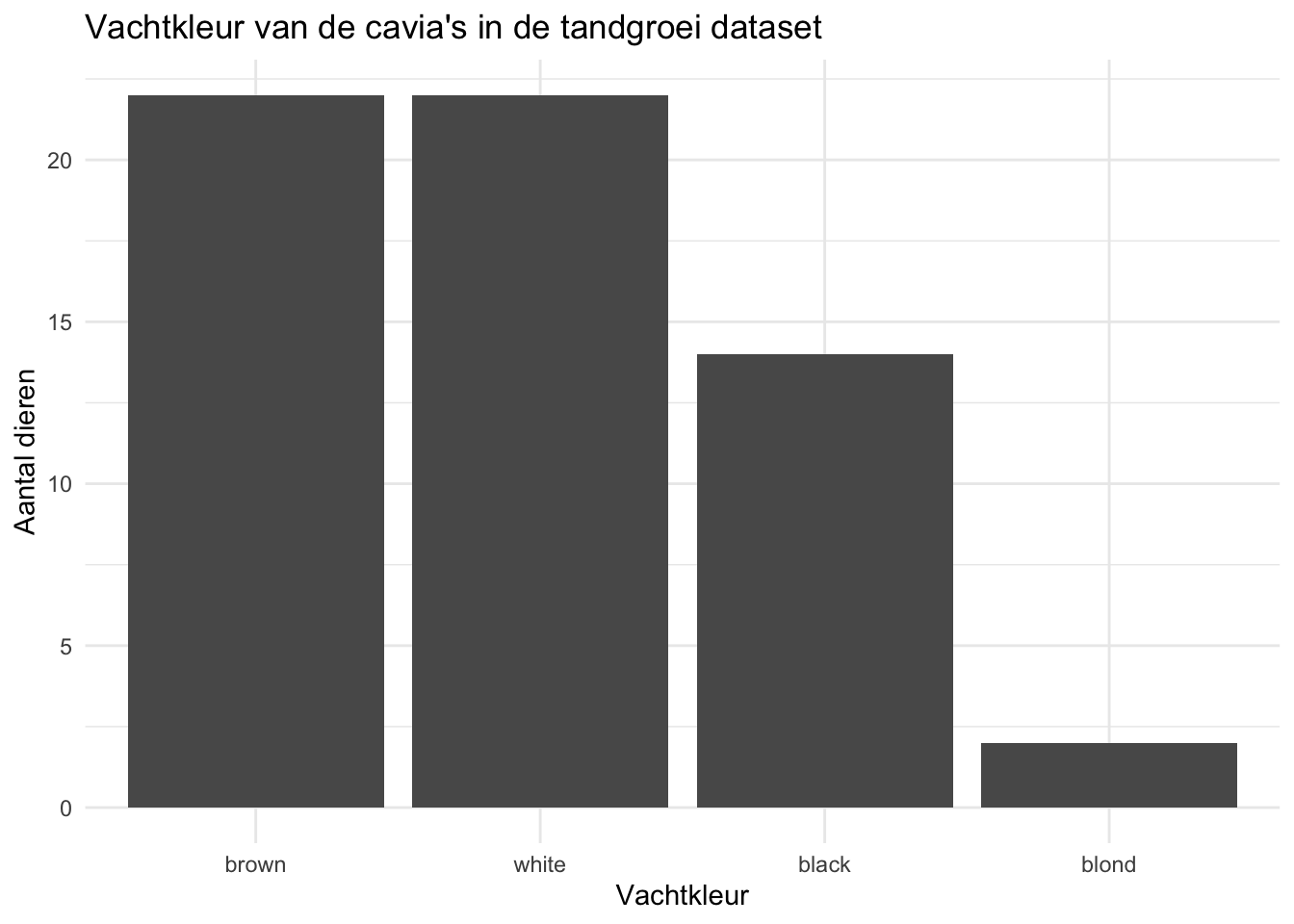
Het is ook nog mogelijk om zelf een volgorde te kiezen. We moeten dan de x-variabele omzetten naar een factor en de levels van de factor aangeven:
tandgroei_compleet |> count(furcolour) |>
ggplot(aes(
x = factor(furcolour, levels = c("black","brown","blond","white")),
y = n)) +
geom_col() +
theme_minimal() +
labs(
title = "Vachtkleur van de cavia's in de tandgroei dataset",
x = "Vachtkleur",
y = "Aantal dieren"
)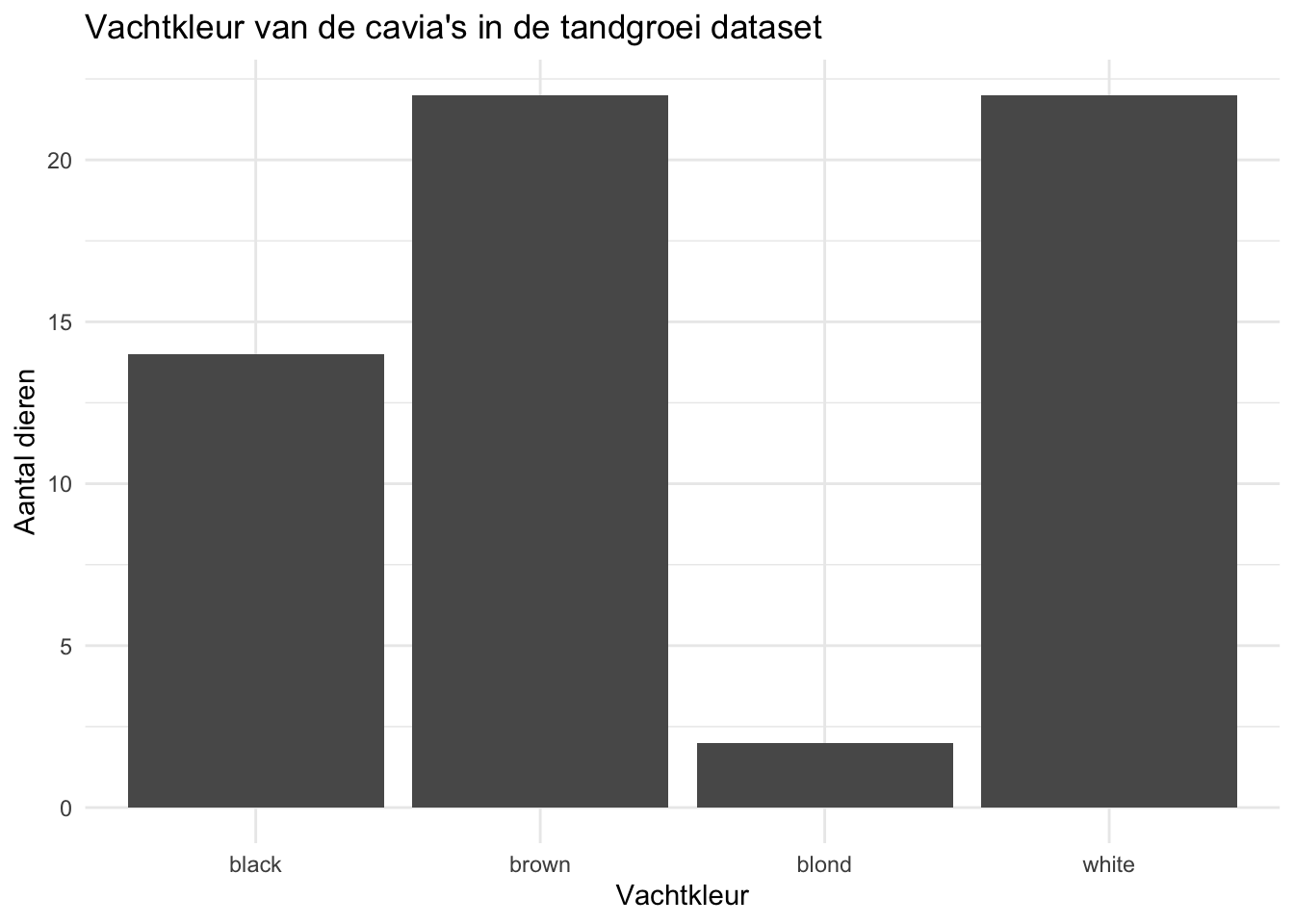
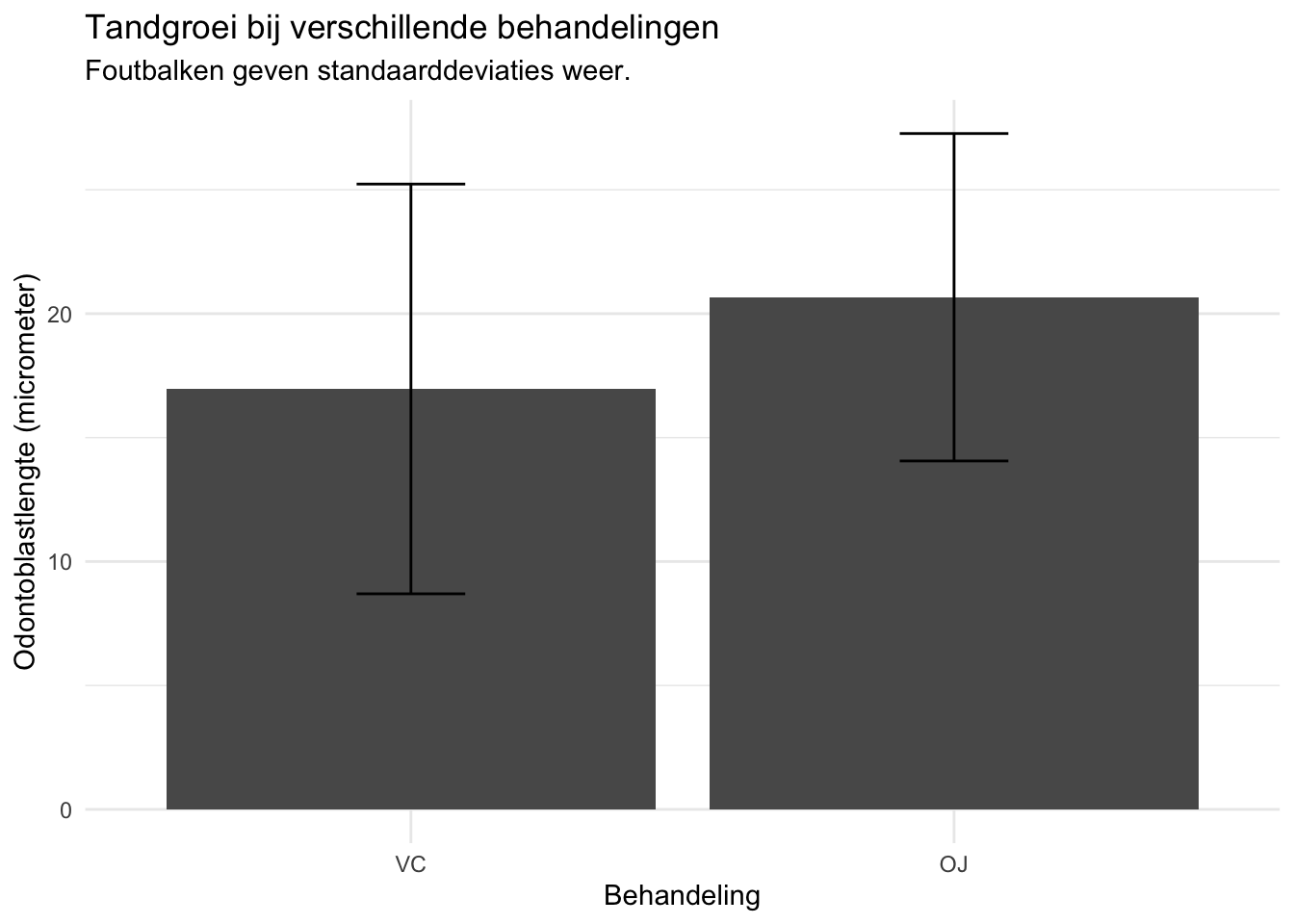
Opdracht 5
Maak voor de tandgroei dataset een staafdiagram met daarin de gemiddelde odontoblastlengte per behandeling (OJ en VC). Zorg dat de staafdiagram voldoet aan het volgende:
- De standaarddeviaties zijn weergegeven als foutbalken.
- De grafiek heeft een informatieve titel en duidelijke aslabels.
- De categorie VC wordt als eerste weergegeven, gevolgd door de categorie OJ.
Klik hier voor het antwoord
tandgroei |> group_by(supplement) |>
summarize(gemiddelde_lengte = mean(length_um, na.rm = TRUE),
sd_lengte = sd(length_um, na.rm = TRUE)) |>
ggplot(aes(
x = factor(supplement, levels = c("VC", "OJ")),
y = gemiddelde_lengte
)
) +
geom_col() +
geom_errorbar(aes(ymin = gemiddelde_lengte - sd_lengte,
ymax = gemiddelde_lengte + sd_lengte),
width=.2) +
theme_minimal() +
labs(
title = "Tandgroei bij verschillende behandelingen",
subtitle = "Foutbalken geven standaarddeviaties weer.",
x = "Behandeling",
y = "Odontoblastlengte (micrometer)"
)
Werkcollege
Casus Les 5
In deze casus gaan we aan de slag met een dataset over hepatitis. De dataset bevat meetwaarden voor 564 proefpersonen. Elk proefpersoon heeft zijn eigen nummer in de dataset (Subject). Verder is voor elke proefpersoon bekend wat het geslacht is (Sex) en wat de leeftijd is (Age). Verder zijn de proefpersonen ingedeeld in drie categorieën (Category):
- Bloeddonoren die gezond zijn (
blood_donor). - Bloeddonoren die mogelijk hepatitis hebben (
suspected_donor). - Hepatitis patiënten (
hepatitis_patient).
Voor alle proefpersonen zijn de bloedwaarden van verschillende stoffen bepaald. De onderzochte stoffen zijn de volgende:
- Albumine (
ALB) in g/L. - Alanine transaminase (
ALT) in U/L. - Aspartate transaminase (
AST) in U/L. - Bilirubine (
BIL) in µmol/L. - Acetylcholesterase (
CHE) in U/ml. - Creatinine (
CREA) in µmol/L. - Gamma-glutamyl transferase (
GGT) in U/L.
De data is verdeeld over twee CSV bestanden. Eén bestand bevat de bloedwaarden voor de bovengenoemde stoffen (casus_les05_metingen.csv) en het andere bestand bevat de informatie over de patiënten (casus_les05_patientinfo.csv).
In deze casus willen we de volgende onderzoeksvragen beantwoorden:
Kan bilirubine gebruikt worden als biomarker voor hepatitis? Met andere woorden: verschilt het bilirubinegehalte in de verschillende patiëntcategoriën?
Kan aspartate transaminase gebruikt worden als biomarker voor hepatitis? Met andere woorden: verschilt het aspartate transaminasegehalte in de verschillende patiëntcategoriën?
Voorbereiding
Open een nieuw script voor je analyses en zorg ervoor dat dit script comments bevat die de stappen in het script beschrijven/toelichten.
Data inlezen
Laad de packages die je wilt gebruiken en lees beide delen van de dataset in in R.
Data inspecteren
- Is er sprake van tidy data? Zo niet, maak de data dan eerst tidy.
- Controleer of er inderdaad 564 patiënten in de dataset aanwezig zijn.
- Bepaal welke categoriën er zijn en hoeveel patiënten er per categorie zijn. Bepaal ook hoeveel mannen en vrouwen er in elke categorie zitten.
Data bewerken
Voeg beide delen van de dataset samen tot één tibble.
Resultaten communiceren
Bepaal voor zowel bilirubine als aspartate transaminase of ze gebruikt kunnen worden als biomarker voor hepatitis. Maak hiervoor zowel boxplots als staafdiagrammen. Zorg ervoor dat de grafieken aan de volgende voorwaarden voldoen:
- Zorg ervoor dat de categoriën op de x-as staan en de volgende volgorde hebben: blood_donor, suspected_donor en hepatitis_patient.
- Zorg voor informatieve grafiektitels en duidelijke aslabels.
Trek conclusies om antwoord te geven op de onderzoeksvragen.