Les 2
Leeruitkomsten
- De student kan een DNA chromatogram en de berekende Q-scores van een Sanger sequencing experiment interpreteren.
- De student kan met behulp van NCBI BLAST mutaties detecteren in een DNA sequentie en bepalen wat het effect van de mutatie is op het eiwit.
Voorbereiding
Biologische achtergrondinformatie
DNA varianten
De nucleotidenvolgorde van een gen (de DNA sequentie) bepaalt de aminozuurvolgorde (en zo de structuur en functie) van een eiwit. In les 1 hebben we gezien dat één gen d.m.v. alternative splicing meerdere transcripten kan voortbrengen en zo verschillende eiwitten (de zogenaamde isoformen). In deze les zullen we zien dat er voor één gen ook verschillende versies van de coderende sequentie kunnen bestaan door de aanwezigheid van DNA varianten.
Alle DNA varianten zijn het gevolg van mutaties. Mutaties zijn veranderingen in de nucleotidevolgorde van het DNA. Sommige van deze veranderingen zijn schadelijk voor het organisme, bijvoorbeeld omdat ze een ziekte veroorzaken. Deze mutaties zullen weinig voorkomen in een populatie. Andere mutaties zijn niet schadelijk voor het organisme en kunnen daarom worden doorgegeven aan de volgende generatie. Op deze manier zullen deze niet-schadelijke DNA varianten in frequentie toenemen.
Een voorbeeld van niet-schadelijke DNA varianten zijn single nucleotide polymorphisms, oftewel SNPs. De biologische variatie tussen individuen van dezelfde soort wordt voornamelijk veroorzaakt door SNPs. SNPs zijn DNA varianten die bij meer dan 1% van de individuen in een populatie voorkomt. SNPs zijn DNA varianten waarbij slecht 1 nucleotide verschilt. Stel bijvoorbeeld dat een specifieke positie in het genoom voor de meeste personen een G is, maar voor een kleine groep een A. Dit betekent dat er een SNP is op deze positie met twee varianten (G en, minder voorkomend, A). SNPs komen erg veel voor in het genoom: gemiddeld is 1 op de 1000 nucleotiden een SNP. Als er een SNP aanwezig is in een gen, kan dit coderen voor een ander aminozuur (missense) of hetzelfde aminozuur (silent).
Een voorbeeld van schadelijke DNA variantie zijn somatische mutaties. Deze mutaties zijn zeldzaam en ontstaan gedurende het leven van een individu in de lichaamscellen. Somatische mutaties worden veroorzaakt door omgevingsfactoren (zoals roken), fouten in het DNA replicatieproces of door pathogenen (bijvoorbeeld virussen). Deze mutaties worden niet doorgegeven aan de volgende generatie, maar kunnen wel leiden tot het ontstaan van kanker. Somatische mutaties kunnen één enkele nucleotide treffen. In dat geval zijn de mutaties silent of missense. Het kan ook zo zijn dat de mutatie een extra stopcodon introduceert (nonsense mutaties), waardoor een korter of niet-functioneel eiwit ontstaat. Ook is het mogelijk dat de mutatie het splicing proces verstoort, wat leidt tot mRNA met missende delen van exonen of juist extra delen van intronen. Hierdoor kan het leesraam van de coderende sequentie verschuiven en kan het eiwit niet goed worden aangemaakt (frameshift mutaties). Ten slotte is het ook mogelijk dat de somatische mutaties leiden tot inserties (toevoeging van extra nucleotiden in het DNA) en deleties (het verwijderen van nucleotiden uit het DNA). Als dit in de coderende sequentie gebeurt kan ook dat leiden tot een verschuiving van het leesraam.
DNA sequencing
Om te bepalen of een gen een SNP of een somatische mutatie bevat, moeten we de DNA sequentie van dat gen bepalen. Hiervoor kunnen we gebruik maken van DNA sequencing. Er bestaan verschillende DNA sequencing technieken. Een van de oudste technieken (en een techniek die nog steeds wordt gebruikt) is Sanger sequencing. Deze methode kan alleen kleine stukken DNA van ongeveer 300-1000 basenparen sequencen.
Voor Sanger sequencing is het noodzakelijk dat je het stuk DNA dat je wilt sequencen eerst amplificeert met PCR. Vervolgens kun je Sanger sequencing uitvoeren met het PCR product en de forward primer die je hebt gebruikt tijdens de PCR (net als PCR heeft Sanger sequencing een primer nodig als start voor de reactie).
De kwaliteit van de Sanger sequencing kan worden weergegeven in een DNA chromatogram. De kwaliteit van de Sanger sequentie is vaak niet goed voor de eerste 15-40 basen, omdat dat de plaats is waar de primer bindt. Verder neemt de kwaliteit af na 700-900 basen.
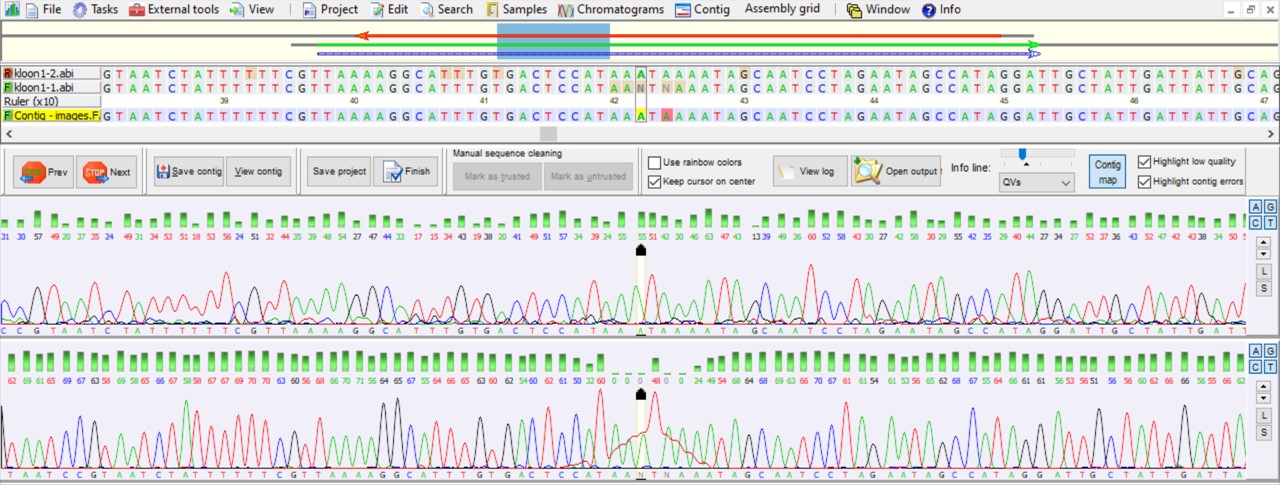
DNA chromatogrammen interpreteren
In het figuur hierboven is een DNA chromatogram weergegeven. Het chromatogram geeft voor elke positie in de DNA sequentie aan welke nucleotiden er op die positie gevonden zijn. Hoe hoger het piekje voor een nucleotide is, hoe vaker die nucleotide op die positie is waargenomen tijdens het sequencen. In het ideale geval heb je dus op elke positie één hoge piek die hoort bij één nucleotide.
In het bovenstaande figuur zie je ook boven elke positie een groen balkje en een getal staan die de kwaliteitsscores weergeven. De formule die hoort bij deze zogenaamde Q-scores is:
\[Q = -10 \cdot log_{10}(E)\] De \(E\) in de formule is de foutkans, dus de kans dat de nucleotide niet correct bepaald is. Deze kans wordt uitgerekend door de software die de Sanger sequencing resultaten bepaalt. Als de kans bijvoorbeeld 0,10 is dat een nucleotide op een positie niet goed bepaald is, dan wordt de Q-score:
\[Q = -10 \cdot log_{10}(0,1) = -10 \cdot -1 = 10\]
Hoe hoger de Q score is, des te betrouwbaarder is de bepaalde nucleotide.
Opdracht 2
- Bepaal de Q-score voor een chromatogrampiek met een foutkans van 0,001.
- Bepaal de foutkans voor een piek met een Q-score van 15.
Klik hier voor het antwoord
- \[-10 \cdot log_{10}(0,001) = -10 \cdot -3 = 30\]
- \[E = 10^{Q/-10} = 10^{15/-10} = 10^{-1,5} = 0,032\]
Opdracht 2
Voor een gezond persoon en twee personen met symptomen van taaislijmziekte werd de DNA sequentie bepaald van een deel van het CFTR gen met Sanger sequencing. De DNA chromatogrammen zijn hieronder weergegeven. Panel A geeft de resultaten weer voor de gezonde persoon. Panel B geeft de resultaten weer voor de patiënten.
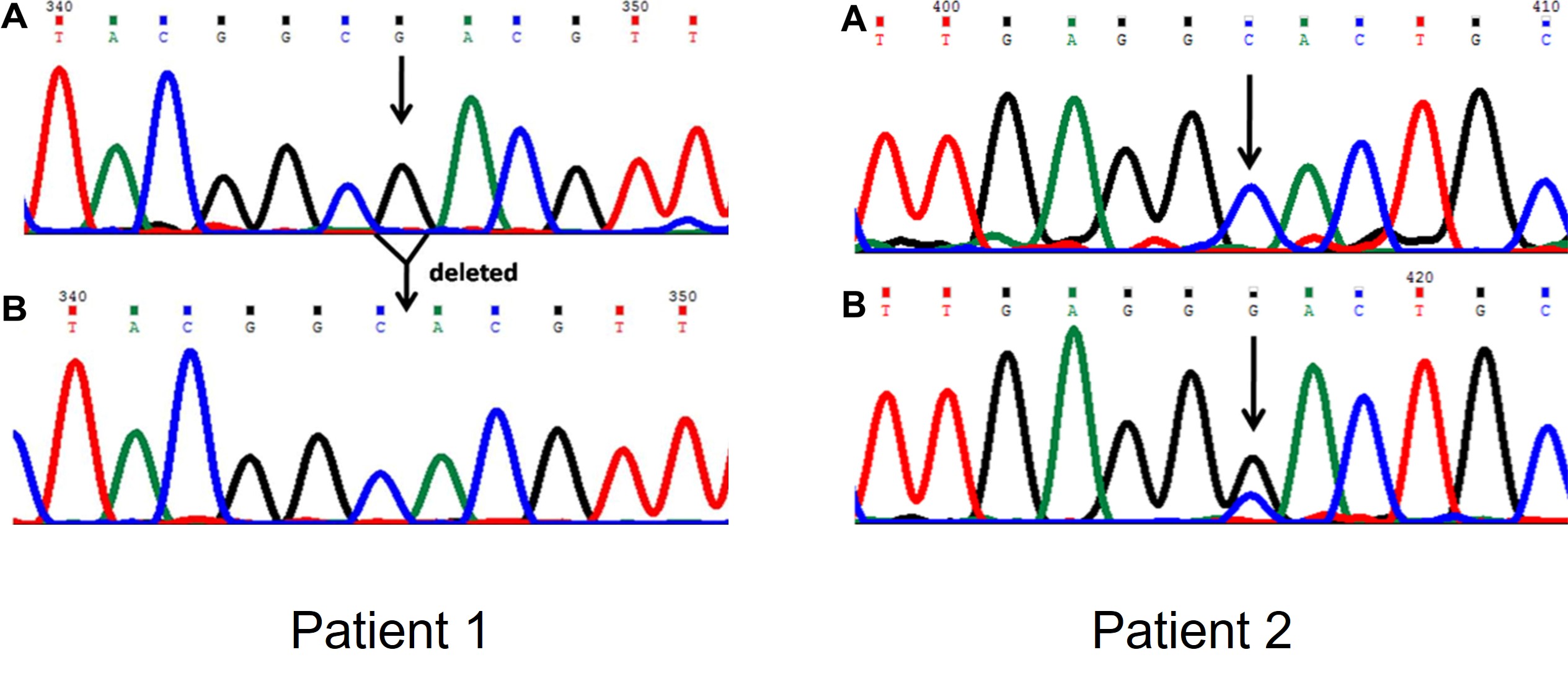
- Wat voor mutatie is er aanwezig in het CFTR gen van patiënt 1?
- Wat voor mutatie is er aanwezig in het CFTR gen van patiënt 2?
- Waarom zijn er twee pieken zichtbaar op positie 405 van de DNA sequentie van patiënt 2?
Klik hier voor het antwoord
- Een deletie.
- Een single nucleotide variant.
- De patiënt is heterozygoot voor de mutatie: slechts één van de kopieën van het gen draagt de mutatie.
DNA sequenties vergelijken met NCBI BLAST
Als je eenmaal de DNA sequentie met Sanger sequencing hebt bepaald, is het niet wenselijk om handmatig een mutatie in de DNA sequentie op te sporen. In plaats daarvan maken we gebruik van de Basic Local Alignment Search Tool, oftewel BLAST. BLAST is een NCBI tool die gebruikt kan worden om DNA sequenties met elkaar te vergelijken. Er zijn twee mogelijke scenario’s voor het gebruik van BLAST.
Scenario 1: een sequentie vergelijken met alle bekende nucleotidensequenties
Je kunt BLAST gebruiken om een DNA sequentie te vergelijken met de miljoenen nucleotidensequenties in de NCBI nucleotide database. Dit is handig als je voor een onbekende sequentie wilt bepalen van welke soort het afkomstig is en van welk deel van het genoom. Ook kun je op deze manier paraloge en orthologe sequenties vinden.
Scenario 2: een sequentie vergelijken met één specifieke andere sequentie
Je kunt BLAST ook gebruiken voor een paarsgewijze vergelijking: in dat geval vergelijk je jouw DNA sequentie met een andere door jouw opgegeven sequentie. In deze les gaan we aan de slag met dit tweede scenario om DNA varianten op te sporen.
Om twee DNA sequenties te kunnen vergelijken, moet BLAST een nucleotide alignment maken. Hiervoor moet BLAST de twee sequenties naast elkaar leggen om te bepalen hoeveel overlap er is tussen de sequenties. Daarbij worden gaten en mismatches toegestaan. Het totale aantal gelijke nucleotiden, de gaten en de mismatches bepalen samen de score van de alignment.
Opdracht 2
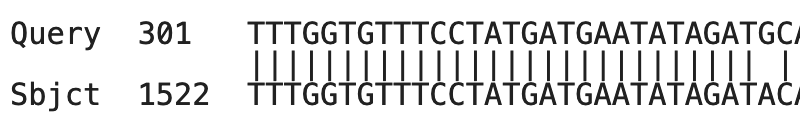
Bepaal voor de onderstaande alignment hoeveel matches, mismatches en gaten er aanwezig zijn:

Klik hier voor het antwoord
Matches = 25
Mismatches = 2
Gaten = 2
Opdracht 2
Maak met de hand (!) een alignment voor de volgende twee sequenties:
TTCCAATTGGCGCGTACATAAA
TTCCTGGCGCGTGCAAAAA
Bepaal ook het aantal matches, mismatches en gaten.
Klik hier voor het antwoord
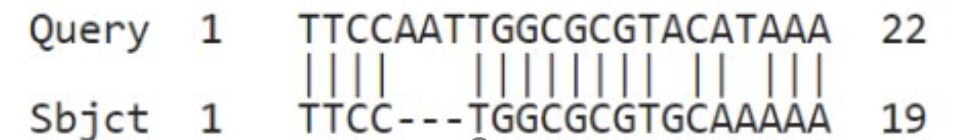
Matches = 17
Mismatches = 2
Gaten = 1
Mutaties detecteren met BLAST
Als je een mutatie wilt vinden met BLAST kun je het volgende stappenplan gebruiken:
Verkrijg de DNA sequentie van (een deel van) het gen met de mutatie. Hiervoor voer je een PCR uit met DNA van de persoon die mogelijk een mutatie heeft. Je gebruikt primers die specifiek de regio van het gen amplificeren waarin de mutatie mogelijk zit (zie de volgende les voor meer informatie over het ontwerpen van primers). Vervolgens gebruik je het PCR product om Sanger sequencing uit te voeren en de DNA sequentie te verkrijgen. De DNA sequentie wordt opgeslagen als een FASTA bestand.
Verkrijg de coderende sequentie (CDS) van het bijbehorende transcript. We willen graag weten of er een mutatie aanwezig is in de door ons bepaalde DNA sequentie en zo ja, wat het effect van deze mutatie is op het eiwit. Hiervoor is het belangrijk om de gevonden DNA sequentie te vergelijken met een referentiesequentie. We gebruiken hiervoor de coderende sequentie van het transcript waarin we geïnteresseerd zijn. In les 1 heb je geleerd hoe je de coderende sequentie voor een transcript kunt verkrijgen uit de NCBI nucleotide database. Soms ben je geïnteresseerd in een specifiek transcript van een gen. Als dat niet het geval is, kun je voor deze stap het eerste transcript gebruiken dat wordt genoemd op de NCBI gene pagina van het gen.
Vergelijk de DNA sequenties met elkaar met BLAST. We kunnen nu BLAST gebruiken om beide DNA sequenties te vergelijken en de mutatie (als die er is) op te sporen:
- Ga hiervoor naar BLAST.
- Vink de optie ‘Align two or more sequences’ aan.
- Vul onder het kopje ‘Enter Query Sequence’ de sequentie in die we hebben bepaald met Sanger sequencing (in FASTA formaat).
- Vul onder het kopje ‘Enter Subject Sequence’ de coderende sequentie in van het bijbehorende transcript (in FASTA formaat).
- Klik op de knop ‘BLAST’ om de twee sequenties te vergelijken.
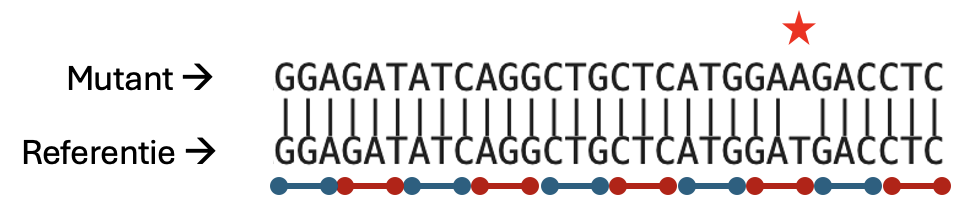
In de onderstaande video worden de bovenstaande stappen uitgelegd aan de hand van een voorbeeld met ACSL1. De in het voorbeeld gebruikte DNA sequentie voor de mutant is te vinden in het bestand human_acsl1_NM_001286708.2_mutant.txt.
In het bovenstaande voorbeeld is een mutatie gevonden op positie 1059 van het CDS van het ACSL1 transcript 2. De sequentie op positie 1059 is veranderd van een T naar een A.
Opdracht 2
Voor drie patiënten met taaislijmziekte zijn de sequenties van het CFTR gen bepaald met Sanger sequencing. De resultaten zijn te vinden in de bestanden human_cftr_NM_000492.4_patient1.txt, human_cftr_NM_000492.4_patient2.txt en human_cftr_NM_000492.4_patient3.txt.
Bepaal voor elk van de patiënten waar de mutatie zit en wat de verandering is. Zoek hiervoor eerst de coderende sequentie op van het CFTR transcript (NM_000492.4).
Klik hier voor het antwoord
Patiënt 1
De mutatie is op positie 1522-1524. Er is sprake van een deletie.

Patiënt 2
De mutatie is op positie 1542 en 1543. Er is sprake van een deletie.

Patiënt 3
De mutatie is op positie 1550. De sequentie is veranderd van een A naar een G.
Bepalen wat de mutatie betekent voor het eiwit
We willen nu graag weten wat het effect is van de gevonden mutatie. We voeren hiervoor de volgende stappen uit:
- Bepaal de positie van de mutatie in het eiwit. In het ACSL1 voorbeeld werd een mutatie gevonden op nucleotidepositie 1059. Omdat nucleotiden in groepjes van drie coderen voor aminozuren (de tripletten/codons), delen we de gevonden nucleotidepositie door drie:
\[1059 / 3 = 353\]
Dit betekent dat de mutatie aanwezig is op aminozuurresidu 353 van het eiwit.
Bepaal het oude en het nieuwe codon dat hoort bij het gevonden aminozuurresidu. De nucleotideposite 1059 is de laatste base van het codon voor aminozuurresidu 353. Positie 1057, 1058 en 1059 vormen dus samen het codon voor aminozuurresidu 353. Het oorspronkelijke codon is dus GAT en het nieuwe coden is GAA.
Bepaal wat de consequenties zijn van de mutatie voor het eiwit. We kunnen met codon tabellen bepalen wat de verandering betekent voor het eiwit. Het oorspronkelijke codon GAT codeert voor asparaginezuur (D). Het nieuwe codon GAA codeert voor glutaminezuur (E). Er verandert dus door de mutatie één aminozuur in het eiwit. Er is daarmee sprake van een missense mutatie.
Opdracht 2
Bepaal voor de drie patiënten met taaislijmziekte uit de vorige opdracht wat de effecten van de mutaties op het CFTR eiwit zijn.
Klik hier voor het antwoord
Patiënt 1
\[ 1522 / 3 = 507 \frac{1}{3} \\ 1524 / 3 = 508 \]
De deletie zorgt ervoor dat het codon voor aminozuur 508 helemaal verdwijnt. Dit codon was TTT en codeert voor fenylalanine (F). De mutatie zorgt er dus voor dat dit aminozuur niet meer aanwezig is in het eiwit.
Patiënt 2
\[ 1542 / 3 = 514 \\ 1543 / 3 = 514 \frac{1}{3} \] Deze mutatie verandert de laatste nucleotide voor het codon van aminozuur 514 en het eerste nucleotide voor het codon van aminozuur 515. De codons waren GAA-TAT en codeerden voor glutaminezuur (E) en tyrosine (R). De codons in de patiënt zijn nu GAA-TAG en coderen voor glutaminezuur (E) en een stopcodon. Door de deletie van de twee nucleotiden ontstaat dus een frameshift en daardoor ontstaat een vroegtijdig stopcodon. Er is dus sprake van een nonsense mutatie.
Patiënt 3
\[1550 / 3 = 516 \frac{2}{3}\] De mutatie verandert het codon voor aminozuur 517 van TAC naar TGC. Hierdoor verandert de tyrosine (R) naar een cysteïne (C). Er is dus sprake van een missense mutatie.
Werkcollege
Casus Les 2
Deel 1
Voor een patiënt is er een vermoeden dat er een mutatie zit in transcript 1 van het ACOX1 gen. Voor deze patiënt is Sanger sequencing uitgevoerd. De gevonden DNA sequentie is te vinden in het bestand casus_les02_human_acox1_mutant.fa.
Beantwoord de volgende vragen:
- Wat is de gen identifier van ACOX1?
- Op welk chromosoom is ACOX1 aanwezig?
- Hoeveel exonen heeft ACOX1?
- In welke weefsels komt ACOX1 (vooral) tot expressie?
- Wat is de functie van het eiwit waar ACOX1 voor codeert?
- Is er een mutatie aanwezig in het ACOX1 gen van de patiënt? Zo ja, wat is die mutatie en wat is het effect van die mutatie op het eiwit?
Deel 2
Voor een andere patiënt is er een vermoeden dat er een mutatie zit in transcript 1 van het CYP3A5 gen. Ook voor deze patiënt is Sanger sequencing uitgevoerd. De gevonden DNA sequentie is te vinden in het bestand casus_les02_human_cyp3a5_mutant.fa.
Beantwoord dezelfde vragen (vraag 1 t/m 6) voor deze patiënt en het gen CYP3A5.