Workshop: Data visualisaties voor RNA sequencing
Introductie
In deze workshop worden drie verschillende grafiektypen behandeld die een belangrijke rol spelen in een RNA-sequencing analyse. Deze grafieken helpen de onderzoeker om kritische te kunnen kijken naar de data en de betrouwbaarheid ervan te kunnen inschatten. Met behulp van opdrachten gaan we aan de slag met de grafieken en leren we meer over hoe de data moet worden geïnterpreteerd.
Veel plezier en succes!
Voorbereiding
Grafieken hebben in wetenschappelijk onderzoek een belangrijke rol. Een grafiek maakt de leesbaarheid van een rapport of artikel aantrekkelijker. Daarbij kan het de kernboodschap van een artikel ondersteunen en kracht bijzetten. In het geval van grote complexe dataset of bij ingewikkelde analyses kan een grafiek de boodschap vereenvoudigen en helpt het de lezer sneller inzicht te krijgen wat de data betekenen. In deze cursus ga je ook aan de slag met grafieken. Je gaat ze maken maar moet ze ook kunnen lezen. Als voor bereiding op de workshop dien je het lesmateriaal van DAVE nog eens door te nemen, specifiek les 2 en les6.
Algemene stappen bij het lezen van een grafiek
- Lees de titel. Wat is het onderwerp en wat is de data afkomst?
- Bekijk de assen. Wat staat er op de as en wat is de eenheid?
- Wat is de schaalverdeling? Is de verdeling gelijk?
- Wat voor grafiek zie je? Neem les6 van DAVE door.
- Is er een patroon te zien in data of valt iets anders op?
- Welke conclusie of boodschap geven de schrijvers mee? Zie titel en bijschrift.
Leeruitkomsten
De leeruitkomsten voor deze workshop zijn hieronder weergegeven en onderverdeeld per grafiek type.
Principle Component Analyse
- De student kan toelichten waarom een Principle Component Analyse wordt uitgevoerd.
- De student kan in grote lijnen uitleggen hoe een Principle Component Analyse werkt.
- De student kan uitleggen aan welke eisen de data voor Principle Component Analyse moet voldoen.
- De student kan de resultaten van Principle Component Analyse interpreteren.
- De student kan uitleggen wat de grafieken van een Principle Component Analyse laten zien.
- De student kan uitleggen wat je met de resultaten van een Principle Component Analyse kan.
Counts & Volcano plots
- De student kan toelichten welke data gebruikt wordt bij het maken van een Count plot, Volcano plot.
- De student kan toelichten waarom een Count plot en een Volcano plot wordt gemaakt.
- De student kan een Count plot en een Volcano plot maken.
- De student kan de resultaten van een Count plot en Volcano plot interpreteren.
Principle Component Analyse (PCA)
In onderzoek wordt er gekeken en gezocht naar correlaties en verbanden. Een bekend voorbeeld van een verband is een ijklijn of kalibratielijn, weergegeven in een scatterplot. De scatterplot illustreert de relatie tussen variabelen. In het geval van een ijklijn, beinvloed de concentratie de waarde van een andere variabele, bijvoorbeeld de extinctie. In deze situatie zijn er twee variabelen (concentratie en extinctie) geplot beide op een eigen as (x-as en y-as) waardoor een 2 dimensionale grafiek ontstaat. In RNA-sequencing worden de expressie waarden van heel veel genen gemeten, elk gen is een variabele. Als elk gen een dimensie voorstelt dan is het al snel niet meer te plotten.
Bij RNA-sequencing wordt een enorme grote dataset gegenereerd met complexe biologische informatie. In deze data zoeken we naar verschillen en overeenkomsten tussen de monsters. Met behulp van een principle component analyse kan het aantal dimensies gereduceerd worden zodat het mogelijk is om patronen te herkenen in de data. Complexe data worden dan vereenvoudigt waardoor het makkelijker is om de data te kunnen interpreteren en conclusies te kunnen trekken. Methoden die de data op deze manier samenvatten worden dimension reduction methods genoemd.
Dit deel van de workshop richt zich op het doorlopen van de stappen van de principal component analyse, en wordt er gekeken naar de resultaten en de interpretatie daarvan. Principle component analyse (PCA) reduceert het aantal variabelen, bij RNA-sequencing het aantal gen dimensies, door er lineaire combinations van te maken. Lineaire combinatie RNA-seq ziet er als volgt uit:
\[4 \times expression\ gene\ A + 2 \times expression\ gene\ B + 6 \times expression\ gene \ C +\ ...\]
In de workshop gaan we niet dieper in op de wiskundige aanpak van PCA, maar bespreken globaal de methode en richten ons vooral op het principe en de toepassing. Een uitgebreidere uitleg over PCA is o.a. te vinden in deze video.
Tijdens de workshop gebruiken we gepubliceerde genexpressie datasets en kijken we naar verschillende type kankercellen.
Om een principle component analyse te kunnen uitvoeren hebben we genormaliseerde data nodig. De datasets waarmee we werken zijn genormaliseerde ruwe counts in de eenheid TPM (Transcript per Million). Alle counts van een gen zijn dan gedeeld door het totaal aantal counts in het monster vermenigvuldigd met een miljoen.
Er zijn verschillende datasets van genexpressie aanwezig van kankertypes, ze staan allemaal in dezelfde folder. Van elk kankertype is een eigen dataset aanwezig. In deze dataset zijn verschillende cellen geanalyseerd van hetzelfde kankertype. Van deze datasets zijn ook combinaties gemaakt van drie kankertypes. Hieronder is uitgelegd hoe je een specifieke dataset kunt inladen.
Van de volgende kankertypes zijn datasets aanwezig.
BR = Breast cancer
CNS = Central Nervous System
CO = Colon
LE = Leukemia
ME = Melanoma
NS = Non-Small lung
OV = Ovarian
RE = Renal
data_CNS = inladen dataset 1 kankertype
data_CNS_ME_CO = inladen van een combineerde dataset van verschillende kankertypes
Deze datasets kunnen geladen worden in R door gebruik te maken van de code in de code chunk hieronder.
# Laad een dataset met genexpressie van verschillende kankertypes
file_naam <- "data_CNS_CO_LE.rds" # Een andere dataset kan geladen worden door een afkorting aan te passen, zie de lijst hierboven.
file_pad <- "/home/data/opra4v/workshop/"
pad_en_naam <- paste0(file_pad, file_naam)
data <- readRDS(pad_en_naam)In de dataset is het aantal read counts uitgedrukt in TPM. Zoals geleerd bij Experimenteel Ontwerp les 5 kun je inzicht krijgen in de verdeling van de datapunten, in dit geval de counts in een monster, door gebruik te maken van de quantile functie, zie hieronder. Bekijk een aantal monsters. Wat valt op?
Neem de log van de read counts en bekijk opnieuw de verdeling van de data. Wat is anders geworden? Voor een snelle analyse van de data kan een log2 worden toegepast. Voor een nauwkeurige analyse kan beter een rlog functie toegepast worden om variaties beter vast te stellen.
Bekijk de dataset. Wat staat er in de headers en wat zijn de rijtitels? Noteer dit in het markdown document. De volgende stap is het uitvoeren van de principle component analyse (PCA).
Na het uitvoeren van een PC analyse kun je de hoeveelheid variatie in elke PC bekijken met de onderstaande code.
Een tabel wordt dan gegeven die een samenvatting laat zien van de variatie in elke PC (1, 2 , 3 etc). Voor een goede analyse van de monsters is het belangrijk om voldoende variatie te plotten die aanwezig is in de data. Een richtlijn is ca 90%. Is de totale variatie in PC1 en PC2 niet toereikent dan dient een tweede plot gemaakt te worden met PC3 of meer.
De variatie, in procenten, kan per principle component geplot worden met de onderstaande code.
# Matrix overzicht PC variatie in object
importance <- summary(pca)$importance
# Vector met PC labels
PC_labels <- paste0("PC", 1:ncol(pca$x))
# Creëer dataframe met PC labels en variatie in %
df <- data.frame(
PC = factor(PC_labels, levels = PC_labels),
variance = importance[2, ] * 100 # som alle variantie van de PC's is 100%
)
# Scree plot (ggplot2) met variatie in %
ggplot(df, aes(x = PC, y = variance)) +
geom_bar(stat = "identity") +
ylab("Verklaarbare variatie (%)") +
xlab("Principal Component") +
ggtitle("Verklaarbare variantie van de Principle components ") +
theme_bw()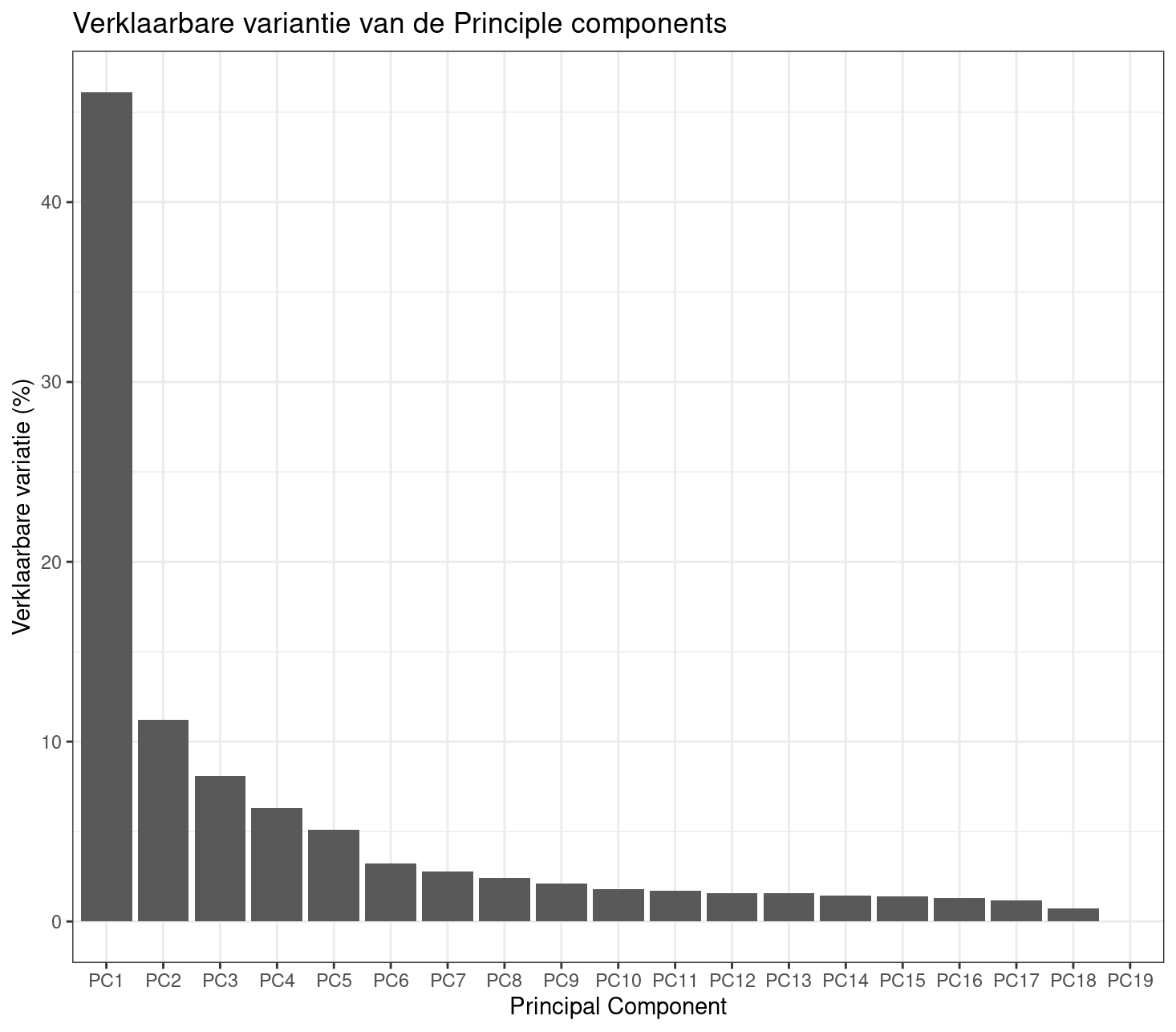
We gaan verder met het plotten van de resultaten van de PCA analyse om te kijken hoe de monsters verdeeld zijn. Om de kankertypes makkelijke te onderscheiden in de PCA plot koppelen we er één kleur aan, met behulp van de onderstaande code.
# Wijs een kleur toe aan de kleuren voor elk kanker type
data_t <- t(data)
data_t <- data_t |>
as_tibble(rownames = "rn") |>
mutate(type = substr(rn, 1, 2))
color_map <- c(
BR = "red",
CN = "steelblue2",
CO = "aquamarine4",
LE = "orange",
ME = "purple",
NS = "cyan",
OV = "pink",
RE = "brown"
)
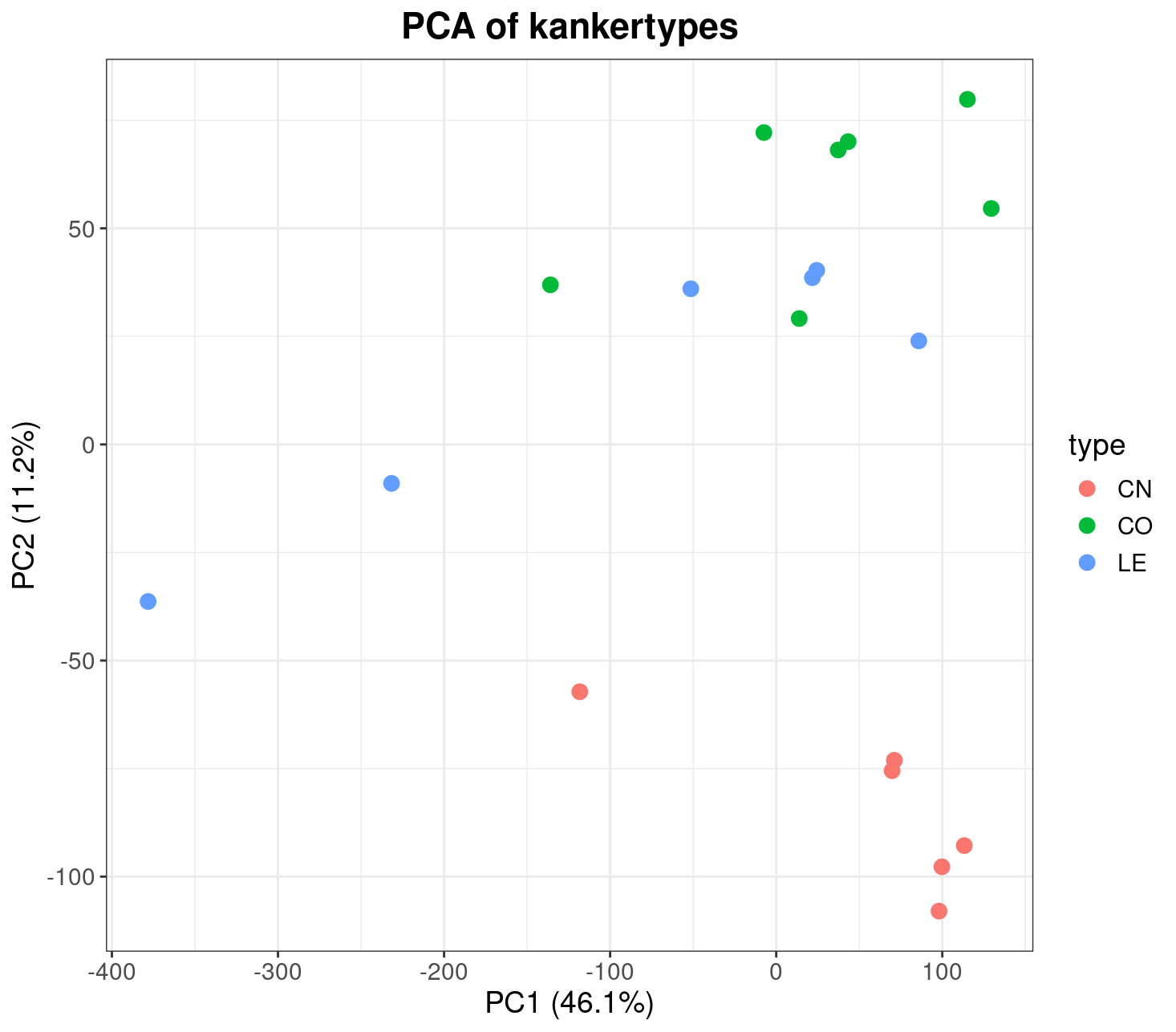
point_colors <- color_map[data_t$type]Nu kunnen de monsters geplot worden in een PCA grafiek. Hieronder een voorbeeld van PC1 tegen PC2.
# Creëer een dataframe met PC waarden
pca_plotting <- data.frame(
PC1 = pca$x[, 1],
PC2 = pca$x[, 2],
type = data_t$type
)
# Voeg een kolom toe met de namen van de monsters
pca_plotting$sample <- rownames(pca$x)
# Maak objecten met de PC waarde
PC1_var <- round(100 * summary(pca)$importance[2, 1], 1)
PC2_var <- round(100 * summary(pca)$importance[2, 2], 1)
# PCA plot (ggplot2) met verklaarbare variantie
ggplot(pca_plotting, aes(x = PC1, y = PC2)) +
geom_point(aes(color = type), size = 3) +
ggtitle("PCA of kankertypes") +
xlab(paste0("PC1 (", PC1_var, "%)")) +
ylab(paste0("PC2 (", PC2_var, "%)")) +
theme_bw() +
theme(
plot.title = element_text(hjust = 0.5, face = "bold"),
text = element_text(size = 14)
)
Opdrachten Principle component analyse
Na de instructie kun je starten met de onderstaande opdracht.
- Start Rstudio en open een nieuw R markdown document.
- Geef jullie namen aan in het markdown document en geef als titel de naam van de workshop.
- Sla het document op met de naam vl4_groepnr_vis_stat.Rmd.
- Lever het markdown document in bij je tutor aan het einde van de les.
- Voeg een code chunck toe aan je markdown document en laad als eerste de benodigde packages.
- Schrijf boven de code chunck toelichting en informatie over de stap die je uitvoert. Dit is natuurlijk belangrijk voor elke chunck en om een begrijpbaar document te krijgen als naslag werk.
- Maak de PCA plot in je markdown document.
Lees de PCA plot af. Wat valt op? Noteer dit in het markdown document!
Hint: Vergeet niet dat het markdown document een naslagwerk is en gebruikt wordt om straks je bevindingen te communiceren aan collega-onderzoekers.
Kies nu zelf een dataset van gecombineerde kankertypes en maak een PCA plot. Als dit lukt vervolg dan met het maken van een PCA plot waar je PC3 en PC4 tegen elkaar uitzet en herhaal dit voor PC5 en PC6. Wat valt je dan op?
Hint: Je kunt ook tekst toevoegen aan elk datapunt zodat je kunt achterhalen waar elk monster in de PCA plot zit. Gebruik eventueel een AI hulpmiddel om dit voor elkaar te krijgen.
Laad nu een dataset met alleen genexpressie van 1 van de kankertypes. Een code voorbeeld is hieronder gegeven. Maak opnieuw een Scree en PCA plot.
Wat valt je op als je naar de PCA en Scree plot kijkt van een dataset van 1 kankertype en dat vergelijkt met een combineerde dataset?
# Laad nu een dataset met genexpressie van 1 van de kankertype uit je eerder gekozen dataset
file_naam <- "data_CNS.rds" # Een andere dataset kan geladen worden door een afkorting aan te passen, zie de lijst hierboven.
file_pad <- "/home/data/opra4v/workshop/"
pad_en_naam <- paste0(file_pad, file_naam)
data <- readRDS(pad_en_naam)Gen Expressie Analyse
De onderzoeksvraag bij differential gene expression is een verschilvraag en gaat dus over het vinden van een wel of geen verschil tussen monsters. Het verschil tussen de monsters wordt bepaald door het experimentele ontwerp van het experiment. Er kan gekeken worden naar verschillende monsters, denk aan een vergelijking tussen lever cellen en long cellen. Een andere veel voorkomende vraag is, of en wat, het effect is van een behandeling op cellen, de cellen zijn dan vaak van hetzelfde type. Het testen van een stof op cellen om te achterhalen of de stof in kwestie een medicinale werking heeft. Echter, de behandeling kan natuurlijk ook iets anders zijn het incuberen bij een andere temperatuur of andere voeding. Om een goed inzicht te krijgen wat het effect is, wordt in RNA-seq analyses gebruik gemaakt van grafieken.
In dit deel van de workshop gaan we aan de slag met de grafieken die toegepast worden om het verschil op RNA niveau te visualiseren.
Een eenvoudige plot die veel gebruikt wordt is de Count plot. Deze grafiek kan bijvoorbeeld toegepast worden om het verschil in ruwe counts voor een gen te tonenen per conditie. Om te oefenen met de grafieken maken we gebruik van een andere dataset. Deze dataset is onderdeel van de R ‘airway’ package). Meer informatie over de data en het onderzoek kun je vinden in het artikel, Himes et al. (2014).
Als eerste laden de natuurlijk de libraries, en maken we een object met de data.
# laad de vereise libraries
library(DESeq2)
library(ggplot2)
# Importeer de data en resultaten
file_data <- "airway_data.rds"
file_results <- "airway_results.rds"
file_pad <- "/home/data/opra4v/workshop/"
pad_en_data <- paste0(file_pad, file_data)
pad_en_results <- paste0(file_pad, file_results)
data <- readRDS(pad_en_data)
results <- readRDS(pad_en_results)Count plot
Deze data bevat veel informatie waaronder gen en expressie informatie met de bijbehorende statistiek. Deze tabel wordt gebtuikt om een count plot te maken. Een count plot maak je tijdens je vooronderzoek om inzicht te krijgen in de data en daarover specifieke vragen te beantwoorden. Alles plotten kan maar dan wordt het erg onoverzichtelijk. Om een overzichtelijke count plot te kunnen maken kan data weggefilterd worden.
Hieronder een voorbeeld om te achterhalen welk gen een significant expressie verschil heeft en het meeste verschil.
# Verkrijg de genen met een significante p-waarde
sign_genes <- results[which(results$padj < 0.05),]
# Verkrijg het referentie id van het meest geupreguleerde gen
topGene <- sign_genes[which.max(sign_genes$log2FoldChange),]
topGene_name <- rownames(topGene)
topGene_nameDe data extraheren de specifieke data van het gen en behandeling uit de dataset.
# Extraheer de waarden uit de dataset voor de ggplot
geneCounts <- plotCounts(data, gene = topGene_name,
intgroup = c("treatment"),
returnData = TRUE)Om dit te visualiseren in een count plot, met ggplot, gebruiken we de onderstaande code.
# Maak een count plot met ggplot
ggplot(geneCounts, aes(x = treatment, y = count)) +
scale_y_log10() +
geom_point(position = position_jitter(width = 0.1, height = 0),
size = 3, colour = "darkgreen") +
xlab("Dexamethasone treatment") +
ylab("Fragment count") +
ggtitle(topGene_name) +
theme_bw()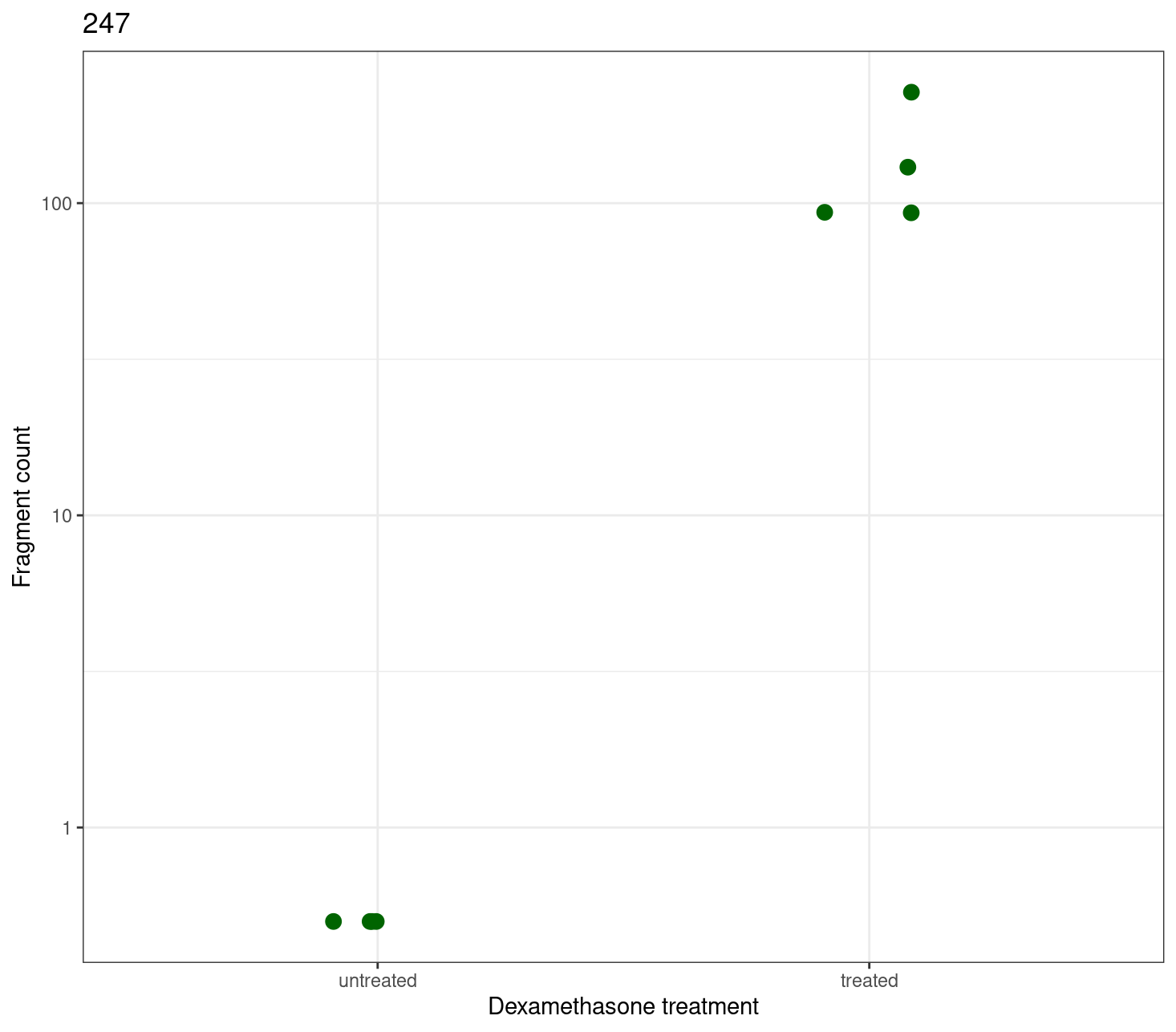
Volcano plot
Om de verandering in genexpressie overzichtelijk en visueel te tonen gebruiken we een Volcano plot. De Volcano plot is instaat de genen te plotten die meer tot expressie komen en significant verschillend zijn op basis van de opgegeven grenzen. Eerst voeren we een filter stap uit.
# Maak een dataframe voor het plotten van genen zonder padj = NA
airway_dge_plotting <- data.frame(results) |> filter(!is.na(padj))
# Voeg een kolom toe waarin is aangegeven of een gen een significante verandering heeft in genexpressie
airway_dge_plotting <- airway_dge_plotting |>
mutate(signif = if_else(padj < 0.05, "padj < 0.05", "Not significant"))Daarna maken we de plot.
# Maak een volcano plot
airway_dge_plotting |>
ggplot(aes(x = log2FoldChange, y = -log10(padj), color = signif)) +
geom_point() +
xlab("log2 fold change") +
ylab("-log10 adjusted p-value") +
ggtitle("Volcano plot - genexpressie, alpha = 0,05") +
theme_bw() +
# Pas de legenda tekst en kleur aan
scale_colour_manual(values = c("grey", "darkgreen"), name = "Significance")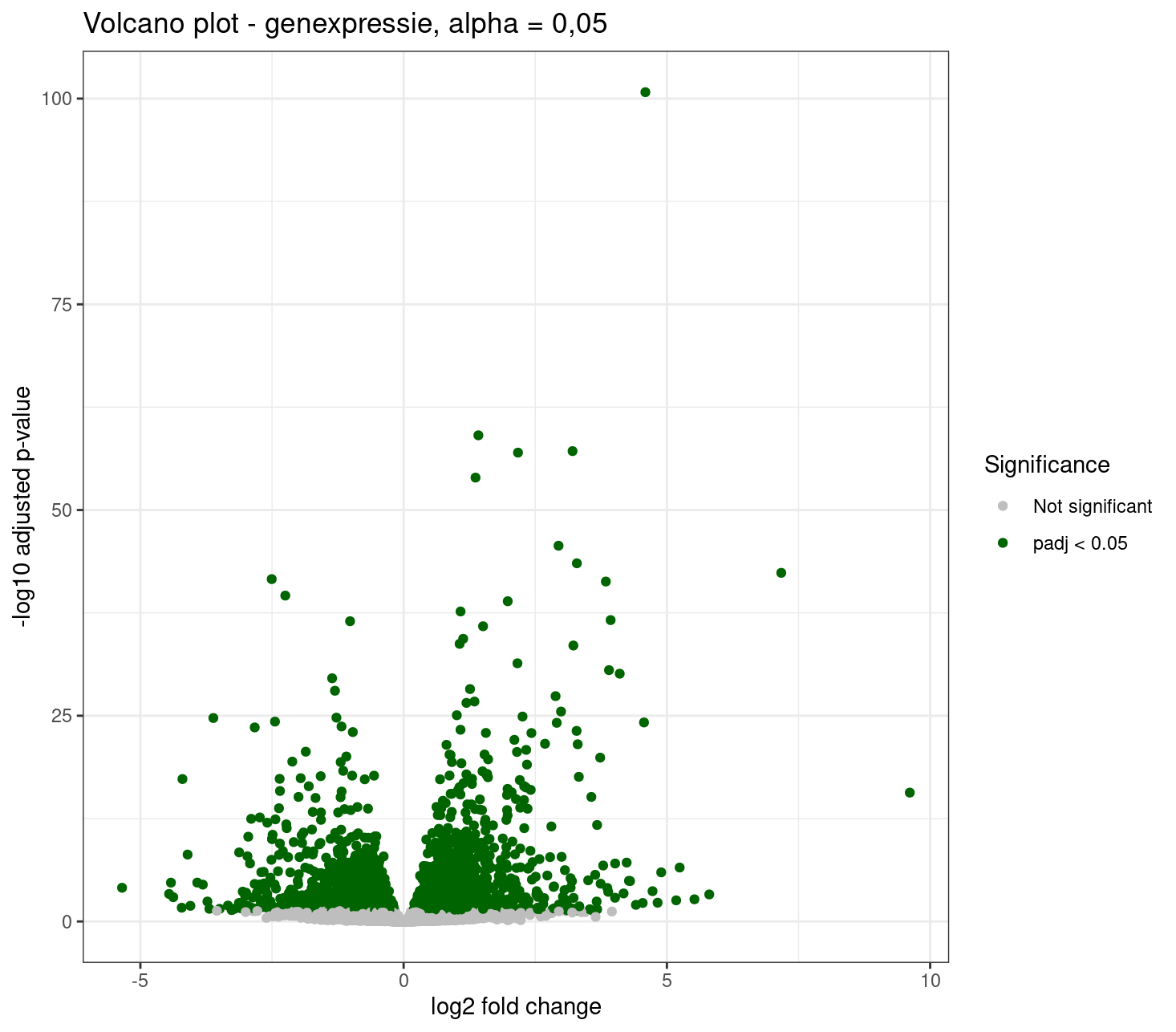
Opdrachten Gen Expressie analyse
Maak de onderstaande opdrachten.
Welke conclusies kun je trekken uit het figuur? Probeer de grafiek aan elkaar uit te leggen.
Hint: Bekijk en lees de grafiek. Gebruik de beschreven richtlijnen die zijn gegeven in de sectie ‘Voorbereiding’.
Pas de code aan waarmee je de grafiek maakt aan zodat je een markering toevoegt aan de grafiek zodat visueel duidelijk is welk gen het meest geupregulateerd is. Markeer met kleur en gen id.
Om te oefenen met het interpreteren en maken van de grafiek gaan we aanpassingen maken in de opgegeven grenzen. We maken een nieuwe Volcanco plot, waarin de kleuren van alle genen met een adjusted p-value < 0.01 and \(|LFC| > 1\) donker rood zijn. Verder willen we een stippellijnen toe voegen met de functies geom_hline() en geom_vline().
Kijk eens of je er uitkomt en maak eventueel gebruik van een AI hulpmiddel.
Welke verschillen neem je waar ten opzichte van de vorige grafiek en wat geven de stippellijnen aan?
Heatmaps
Het laatste grafiek type dat behandeld wordt, is de heatmap. Een heatmap maakt gebruik van kleuren om overeenkomsten en verschillen zichtbaar te maken. Je kunt een heatmap zien als een matrix van getallen binnen een bepaald bereik, waarbij een kleurenschaal deze waarden omzet in kleuren. De kleur van een cel in de matrix wordt dus bepaald door de onderliggende numerieke waarde. Ter verduidelijking: een voorbeeld van een kleurenschaal is wanneer de laagste waarde donkerblauw wordt weergegeven en de hoogste waarde donkerrood. Zo’n schaal helpt om snel patronen en clusters in de data te herkennen. Er is bovendien veel vrijheid in de keuze van kleuren. Onze hersenen zijn goed in het waarnemen van subtiele kleurverschillen wanneer kleuren direct naast elkaar liggen. Op grotere afstand of bij niet‑aangrenzende kleuren wordt het echter moeilijker om de intensiteit nauwkeurig in te schatten en vast te stellen of tinten gelijk zijn.
Een voorbeeld van een heatmap is hieronder gegeven waarbij de toename van de concentratie PFAS is uitgezet tegen het effect op de wormen.
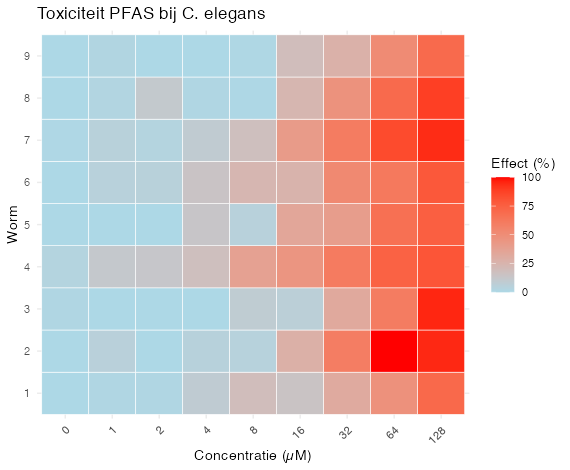
Bij RNA-seq analyses kan de heatmap toegepast worden om inzicht te krijgen in de correlaties en relatieve toename in genexpressie. Een belangrijke controle stap is bijvoorbeeld om te kijken hoeveel de monsters op elkaar lijken die je onderzoekt en hoe zij groeperen. Uiteindelijk is het voor de genexpressie analyse interessant om ook zicht te krijgen op de variatie van expressie bij de monsters en hoe ze groeperen. Hieronder is daar een voorbeeld van gegeven.
Opdrachten Heatmaps
Bekijk de grafiek hierboven. Bespreek je waarnemingen en schrijf deze op in het markdown document. Wat is je conclusie?
Heatmaps vergemakkelijken het vinden van overeenkomsten. In de heatmap hieronder is de correlatie tussen de monsters weergegeven. Monsters die op elkaar lijken worden bij elkaar geplaatst en dus geclusterd. Een veelgebruikte methode van clustering, is hiërarchisch clusteren. Daarbij wordt een zogenoemd dendrogram gemaakt: een boomdiagram dat je naast de rijen en kolommen in de heatmap ziet. Zo’n dendrogram wordt stap voor stap opgebouwd. Eerst wordt gekeken welke twee samples het meest op elkaar lijken. Daarna wordt het volgende meest vergelijkbare sample toegevoegd. Zo gaat het proces door totdat alle samples in de boom zijn opgenomen. Om de gelijkenis tussen twee samples te bepalen, gebruikt de functie pheatmap() de Euclidische afstand, die je kent uit de stelling van Pythagoras.
Om meer ervaring op te doen met het aflezen van zo’n heatmap gaan we aan de slag met de opdracht hieronder.
# Importeer de data
file_metadata <- "airway_metadata.rds"
file_data2 <- "airway_data2.rds"
file_pad <- "/home/data/opra4v/workshop/"
pad_en_metadata <- paste0(file_pad, file_metadata)
pad_en_data2 <- paste0(file_pad, file_data2)
metadata <- readRDS(pad_en_metadata)
data2 <- readRDS(pad_en_data2)
# Maak heatmap
pheatmap(data2, annotation = metadata["treatment"], main = "Clustering van glucocorticoide behandelde spiercellen")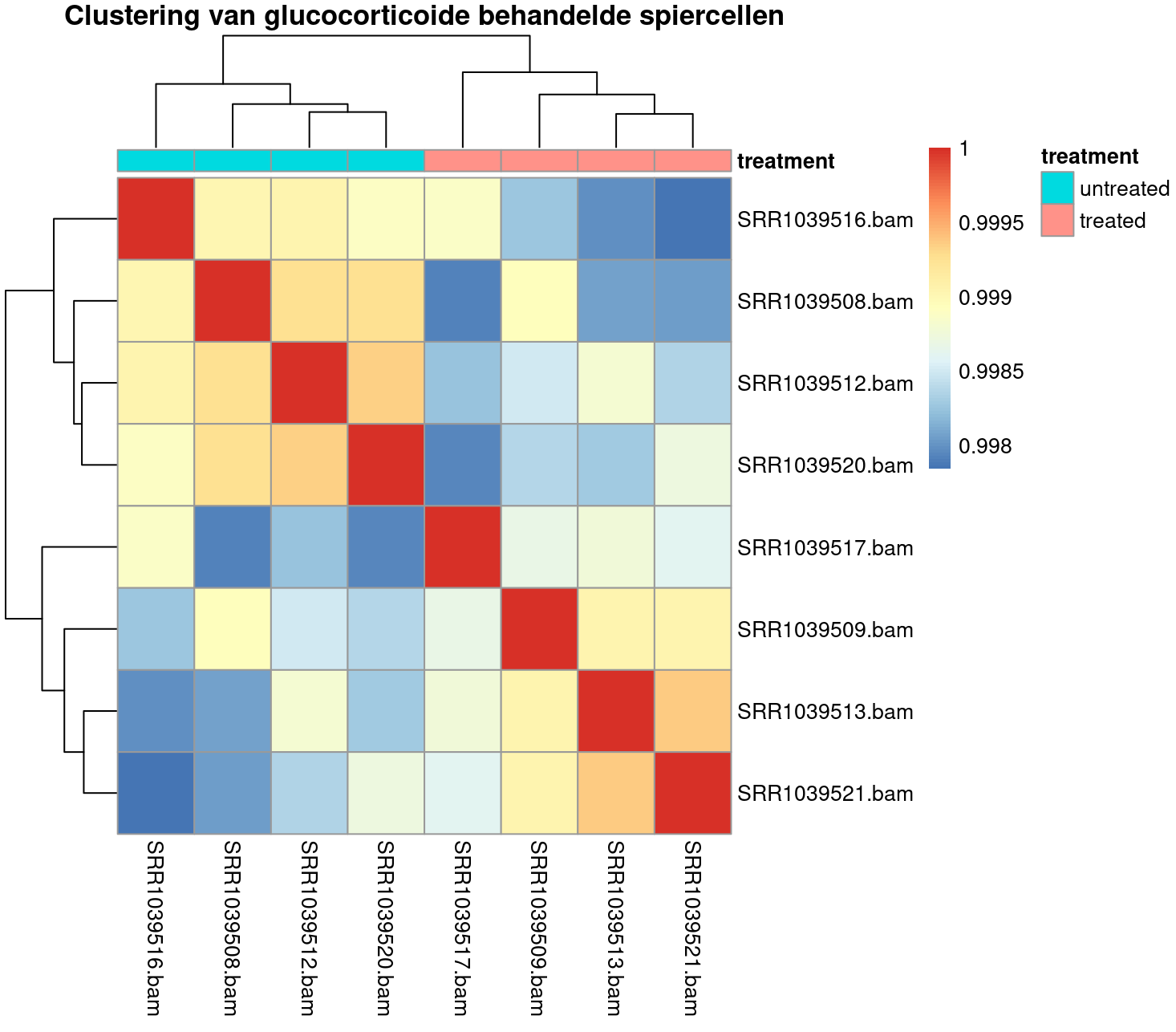
Bekijk de grafiek hierboven. Bespreek je waarnemingen en schrijf deze op in het markdown document. Wat is je conclusie?
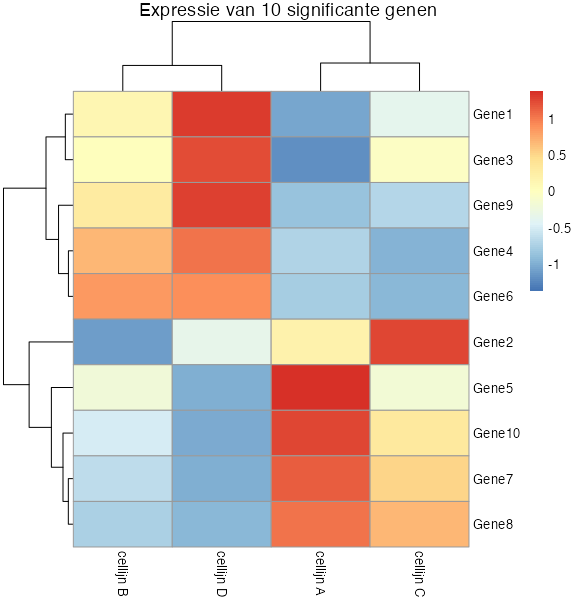
Met de onderstaande code kan een heatmap gemaakt worden waarbij we gebruiken maken van de airway dataset. We plotten de 10 meeste significante genen waarbij we gebruik maken van de absolute waarden die gemeten zijn.
# Pak de 10 meest significante genen.
top10_genes <- rownames(results[order(results$padj)[1:10],])
# Pak de count waarden van de genen
count_values <- assay(data)[top10_genes,]
# Vervang de kolomm met namen met condities
colnames(count_values) <- colData(data)$treatment
# Visualiseer de resultaten in een heatmap
pheatmap(count_values, show_rownames = TRUE, main = "Expressie van de 10 meest significant genen")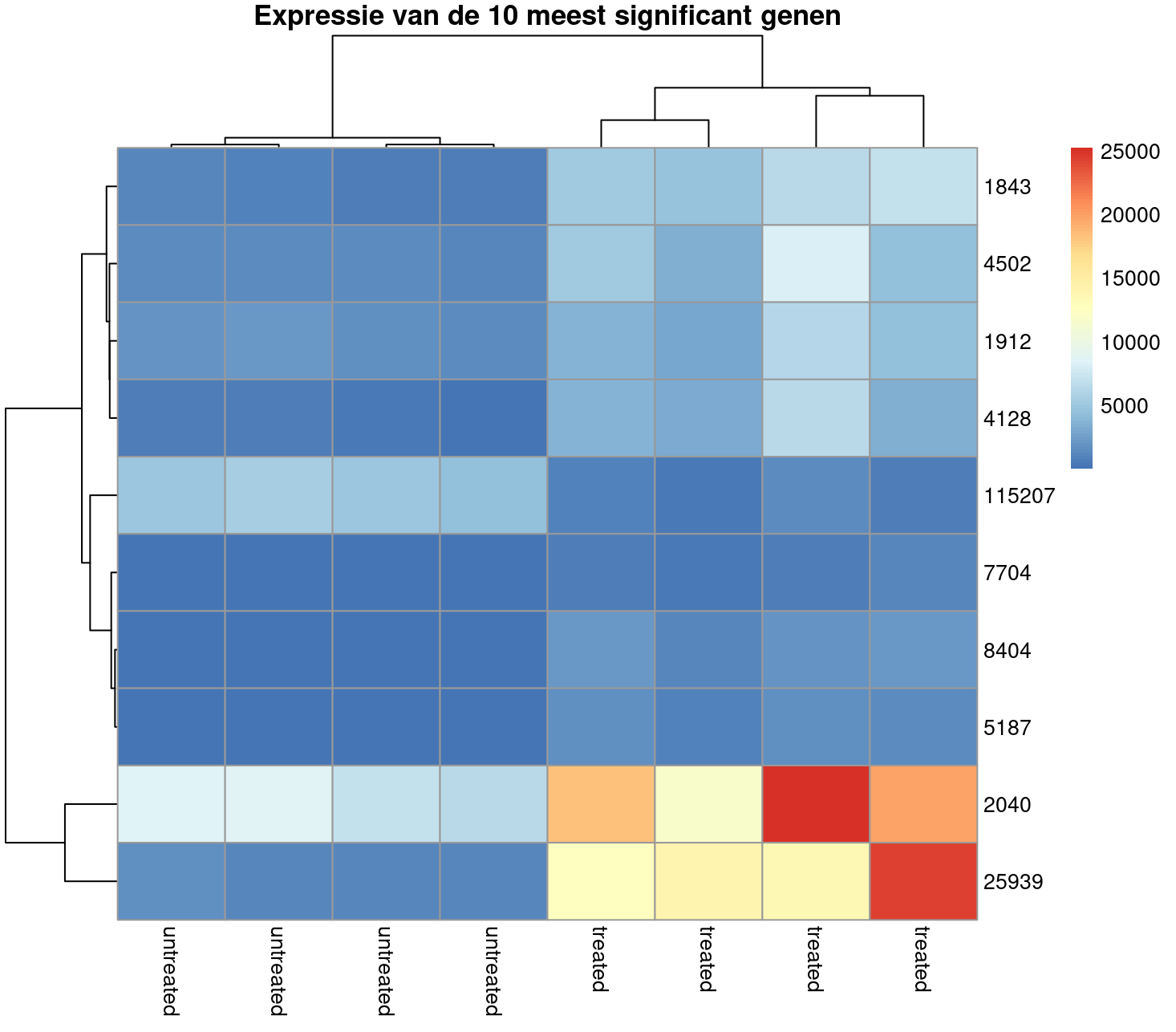
Als het belangrijk is om het verschil in expressie goed in de grafiek naarvoren te laten komen, kunnen we gebruik maken van de schaal optie.
Maak nu zelf de heatmap waarbij je de scaling toepast per rij.
Welke genen zijn het meeste geupreguleerd en gedownreguleerd? En, had je deze conclusie ook kunnen trekken op basis van de niet geschaalde heatmap?