Lesson_04 - Genes
Learning objectives
The student is able:
- To explain the different elements of premature and mature mRNA
- To obtain gene information using the NCBI gene database
- To obtain sequence information and FASTA sequences using the NCBI nucleotide database
- To analyse gene information using bash
Features of Genes
In the previous lesson we have learned to use the KEGG database to gather information about phenotypes. KEGG maps provide an overview of genes and their relations in a biological process. To understand the precise molecular role of a gene in a biological process we have to gather information about the gene itself.
Interesting descriptive research questions are:
- Where is the gene located in the genome?
- What is the size of the gene?
- What is the DNA / RNA sequence of the gene?
- How many exons does the gene have?
- What is the size of transcripts / coding regions?
- Are there mutations in the gene that causes disease phenotypes
- Does the gene have homologs:
- ortholog genes (after a speciation event)
- paralogs genes (after a gene duplication event, within the same species)
- ortholog genes (after a speciation event)
- Does this gene have different alleles (think about Mendel or the different blood types)
- How many transcripts are transcribed from the gene?
- Where en when is the gene expressed and at what level?
- What is the biological function of the gene product?
In this lesson we will try to answer some of the above questions.
NCBI
A starting point to gather information on genes is the NCBI database. The NCBI database consists of numerous databases related to genomes, genes, proteins, diseases and so on (Figure 39). All these database within NCBI are link together and also with other external databases (relational databases)
Figure 39: NCBI database
NOTE: There are numerous other databases with human gene information:
https://medlineplus.gov/genetics/gene
https://www.genecards.org/
https://www.omim.org/
NCBI gene database
To obtain information of a particular gene, we will use the NCBI gene database. In the previous lesson we have studied the molecular pathways of colorectal and gastric cancer and observed that the human Smad2/3 complex acts as an tumorsuppressor complex in colorectal cancer:
- Select the gene database in the dropdown menu (Figure 39)
- Type Smad2 and press enter (or any other gene name you’re interested in)
- Click on the link of the human Smad2 gene
On the right hand side is a “Table of contents” for easy navigation: it provides a shortcut to different sections of information on the gene of interest:
NCBI gene: summary
lesson_04_assignment
- What is the gene ID of the human Smad2 gene?
(Gene IDs are very useful because a gene name can be similar in different species whereas is unique)
Read the Summary of the Smad2 gene page
- In which molecular processes does the Smad2 gene play a role?
At the end of the general “Summary” page we see the option Orthologs.
- Click on the link “all” to view vertebrate orthologs
We observe that the human Smad2 gene has 336 vertebrate orthologs. The smad2 gene is evolutionary well conserved (more on that in lessons to come)
NCBI gene: Genomic regions, transcripts, and products
- Navigate to “Genomic regions, transcripts, and products”
The complete gene (including introns) is depicted with a horizontal green line. The exons are depicted as vertical green boxes (they are appear as a vertical line because the size is small relative to the whole gene). The human Smad2 gene produces 3 transcripts. The transcripts are spliced and translated into proteins. Each transcript has its own unique identifier (red circle) starting with NM_ (the second letter M stands for mRNA). On the right hand side are the corresponding protein identifiers (purple circle) which starts with NP_ (the second letter P stands for protein) (Figure 40). If you point with your cursor to the different transcripts additional information will pop up.
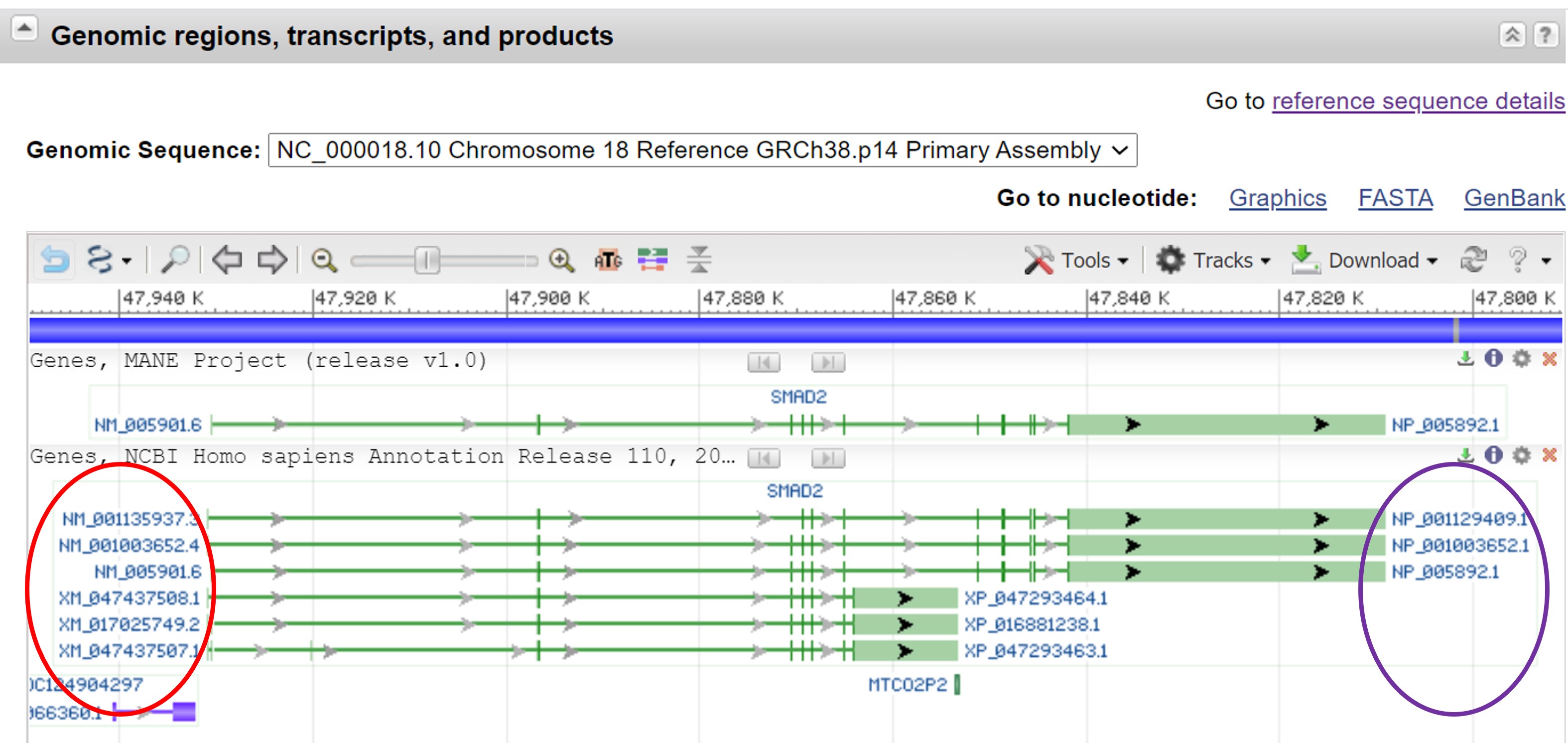
Figure 40: NCBI database: Genomic regions, transcripts, and products
lesson_04_assignment
What is the protein length of mRNA of transcript NM_005901.6?
IMPORTANT: All known transcripts and translated proteins have a unique NCBI identifier. If the identifier is known, use the identifier to retrieve information from the NCBI databases. Other databases (ensembl, UCSC, uniprot) have there own identifiers. Generally, if you refer to a nucleotide- or protein sequence make use of the unique identifier.
NCBI gene: Expression
- Navigate to “Expression”
The section “Expression” shows a list of tissues where the gene is expressed (Figure 41). Be aware that there are different sets of expression data to choose from. These expression profiles are based on RNA-seq. The level of expression is expressed as RPKM. Expression levels can range from 0 (not expressed) to > 100 RPKM (expressed at high levels)
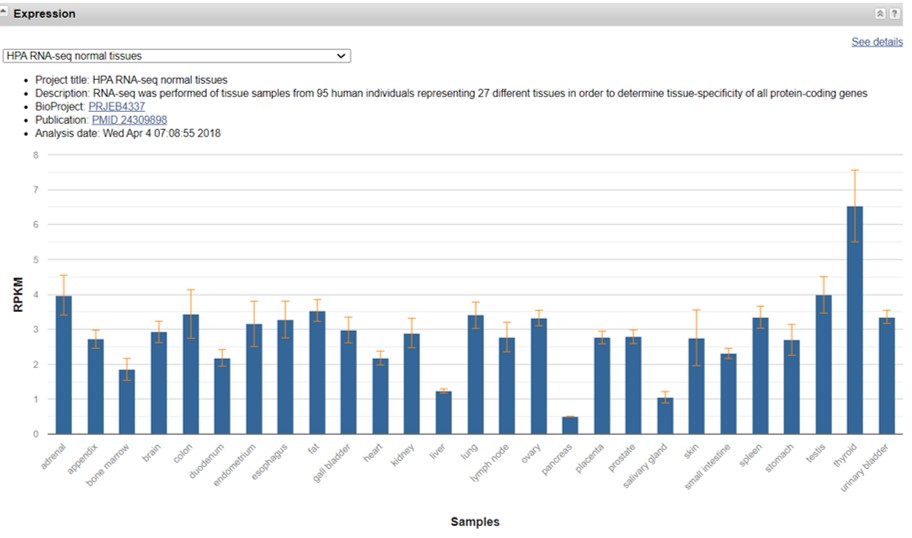
Figure 41: NCBI database: expression
We observe that Smad2 is broadly expressed at low levels in a variety of tissues. If you point with your cursor to the expression bar the exact expression levels will be shown.
lesson_04_assignment
Smad9 is part of the Smad family of genes and interacts with Smad4
- in which tissue does the Smad9 gene have the highest expression?
- What is the expression level of Smad9 in the this tissue?
- Gene expression values are expressed as RPKM. Google this term and explain this unit
NCBI gene: General gene information
- Navigate to “General gene information”
The section “General gene information” shows information about Homology and Gene Ontology
- Click on the Gene Ontology link
We see a description of the function of the Smad2 gene based on three categories: Function, Process, Component.
lesson_04_assignment
The component section states that the Smad2 complex is present in the cytosol, nucleus and chromatin. Can you provide an explanation why this protein is present in three different cellular location.
HINT: What type of protein is Smad2 (look in the Function section)
NCBI nucleotide database
To obtain the mRNA sequence we make use of the NCBI nucleotide database. This database contains hundreds of millions of nucleotide sequences (DNA / RNA) from a diverse set of organisms. Each sequence present in the NCBI nucleotide database has a unique identifier. This database can be accessed directly form the gene information page:
- Go to the “Table of contents” side bar on the right
- Navigate to “NCBI Reference Sequences (RefSeq)”
- Click on the link of the mRNA identifier NM_005901.6 (3rd transcript)
Alternatively, go directly to the NCBI nucleotide database and type the identifier NM_005901.6
We are now in the NCBI nucleotide database (Figure 42)
Figure 42: NCBI nucleotide database
IMPORTANT: Before we start retrieving nucleotide sequences it is important that we understand the different functional elements of eukaryotic mRNA. When a protein coding gene is transcribed it produces precursor mRNA (pre-mRNA). Pre-mRNA consists of different functional elements (Figure 43)
- 5’UTR: untranslated region
- Start codon: ATG, signals the beginning of translation of the transcript sequence
- Exon: part of mature mRNA after splicing
- Intron: non-coding sequence of a transcript that is removed after splicing
- Stop codon: TAG, TGA of TAA, signals the end of translation
- 3’UTR: untranslated region
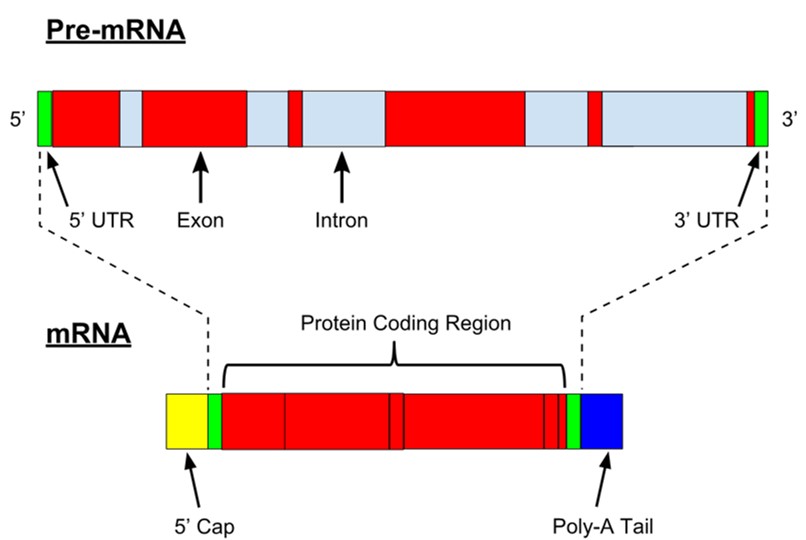
Figure 43: Structure of mRNA
The precursor mRNA is subsequently processed into mature RNA by splicing and the addition of a 5’CAP and a poly-A-tail (Figure 43). The exons that code for the protein are collectively named the “protein coding regions” or coding sequence (CDS). The CDS of a gene always start with a startcodon (ATG) and ends with a stop codon (TAA, TAG, TGA)
If we want to select a specific part of the mRNA transcript we have to scroll down until the FEATURES section appear on the left hand side (Figure 44)
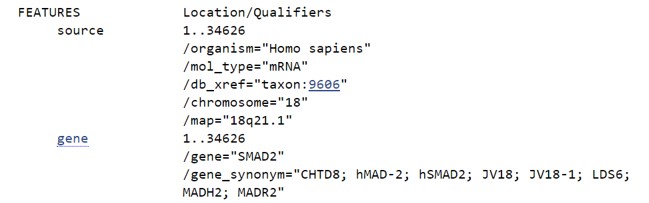
Figure 44: NCBI nucleotide FEATURES section
The FEATURES section contains information about the transcript.
- What is the size of the mRNA
- How many exons are present in the mRNA
- The coordinates of the exons in the mRNA
- The coding sequence (CDS).
- Click on gene (Figure 44)
- Open the drop down menu in the left lower corner to select a specific part of the mRNA sequence (Figure 45)
After selection, in the bottom bar you can click on Details to see more information on this part of the mRNA sequence (it should already be present) or click on FASTA to obtain the sequence (Figure 44)
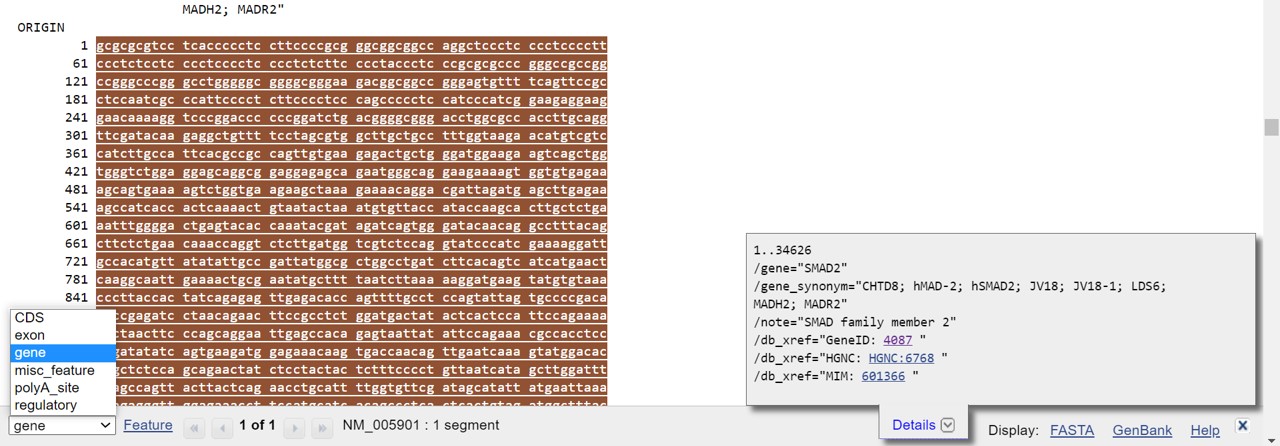
Figure 45: FEATURE drop down menu
IMPORTANT: The general format to store nucleotide / amino acid sequences is FASTA format. FASTA format means that the first line of the sequence starts with a > sign optionally followed with information about the sequence: for example the name of the identifier, species, genome coordinates of the regions and so on (see example below). The second line starts with the actual sequence.
>NM_005901.6 Homo sapiens SMAD family member 2 (SMAD2), transcript variant 1, mRNA GCGCGCGTCCTCACCCCCTCCTTCCCCGCGGGCGGCGGCCAGGCTCCCTCCCCTCCCCTTCCCTCTCCTCCCCTCCCCTCCCCTCTCTTCCCCTACCCTCCCGCGCGCCCGGGCCGCCGGCCGGGCCCGGGCCTGGGGGCGGGGCGGGAAGACGGCGGCCGGGAGTGTTTTCAGTTCCGCCTCCAATCGCCCATTCCCCTCTTCCCCTCC
lesson_04_assignment
- What are the coordinates of the CDS of the Smad2 gene?
- What are the coordinates of the first and the last exon of the Smad2 gene
- What do you notice about the first and the last exon relative to the CDS?
lesson_04_assignment
- Use the NCBI nucleotide database to obtain the FASTA sequence of the CDS of the human Smad2
- Copy the sequence to either notepad (kladblok) or textedit (Mac)
- Save the file as lesson_4_opdracht6_NM_005901.fa
- Move the file to the appropriate folder in your home directory on the Rstudio server
(we will use this sequence later in the course)
Bacterial genes
In the previous lesson we have learned that the bacteria Helicobacter pylori plays a role in gastric cancer. One of the most critical concerns in health science is antimicrobial resistance (AMR) which is the ability of microorganisms to withstand antimicrobial treatments. AMR is also increasing in Helicobacter pylori and to understand / fight AMR we have to understand the mechanism behind AMR. The first start is to study genes that can possibly neutralize the antibiotics. For example genes could act as bacterial pumps to secrete the antibiotics or acts as enzymes that metabolize the antibiotics to a less effective substance. Here, we will use the penicillin binding protein (pbpA) as an example of a gene that increases AMR in bacteria.
To retrieve information on bacterial genes we make use of the nucleotide database. Remember that bacterial genes don’t have introns so there are no alternative transcripts. Also there is only one type of cell where the gene is expressed, the bacteria itself. Bacteria are easily sequenced because of their small genomes and therefore thousands of complete genomes are available in this database.
- Go to the NCBI nucleotide database
- Search for the complete genome of Helicobacter pylori (Figure 46)
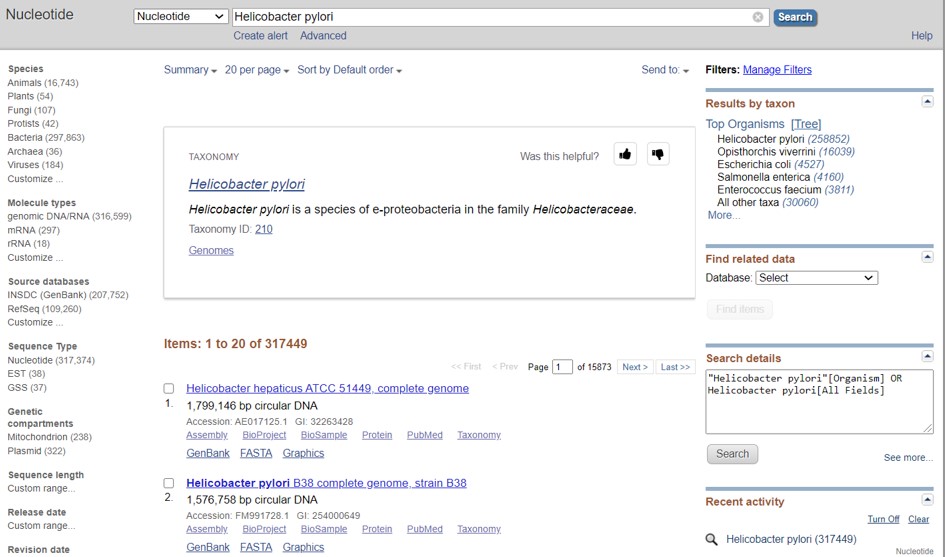
Figure 46: NCBI nucleotide bacterial genomes
- Select strain B38 (Figure 46)
The whole bacterial genome including description of all genes should be loaded into the webpage. This takes a few seconds. If this is not happening go to “Customize view” and select “All features” -> “Update View”. Within the webpage you can now search for your gene of interest by using the search option of your webbrowser (ctrl f) and the name of your favorite gene:
- Search for pbpA (Figure 47)
- Click on the CDS link
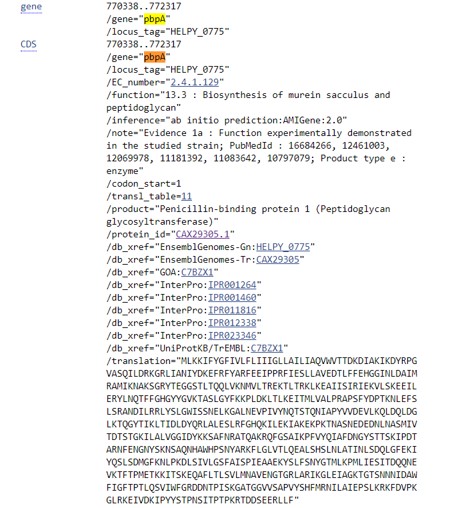
Figure 47: Bacterial genomes gene information
lesson_04_assignment
Use the NCBI nucleotide database to obtain the FASTA sequence of the CDS of the Helicobacter pylori pbpA gene
Save the FASTA as lesson_4_opdracht7_hpylori_pbpa.fa and store on the server (we will use this sequence later in the course)
What is the protein_id of the Helicobacter pylori pbpA gene?
What is the function of this gene?
In the previous assignment we search with ctrl f within the webbrowser for the pbpa gene. But what if we want to systematically search the whole genome for genes or description of genes such as function or enzyme number (=EC_number) and so on.
It would be better to retrieve all the genome information, upload the file to the server and use bash commands to retrieve any information we are interested in.
To obtain all the genome information of Helicobacter pylori in a text file:
- Click on “Send to” (Figure 48, red rectangle)
- Click on file (Figure 47, purple rectangle)
- Choose Genbank in the dropdown menu
- Click on “Create file” and the file will be downloaded to your computer
- Rename the file lesson_4_opdracht8_Hpylori_sequence.gb
(The extension gb means the data is derived from the NCBI genbank database)
- Upload the file to the server
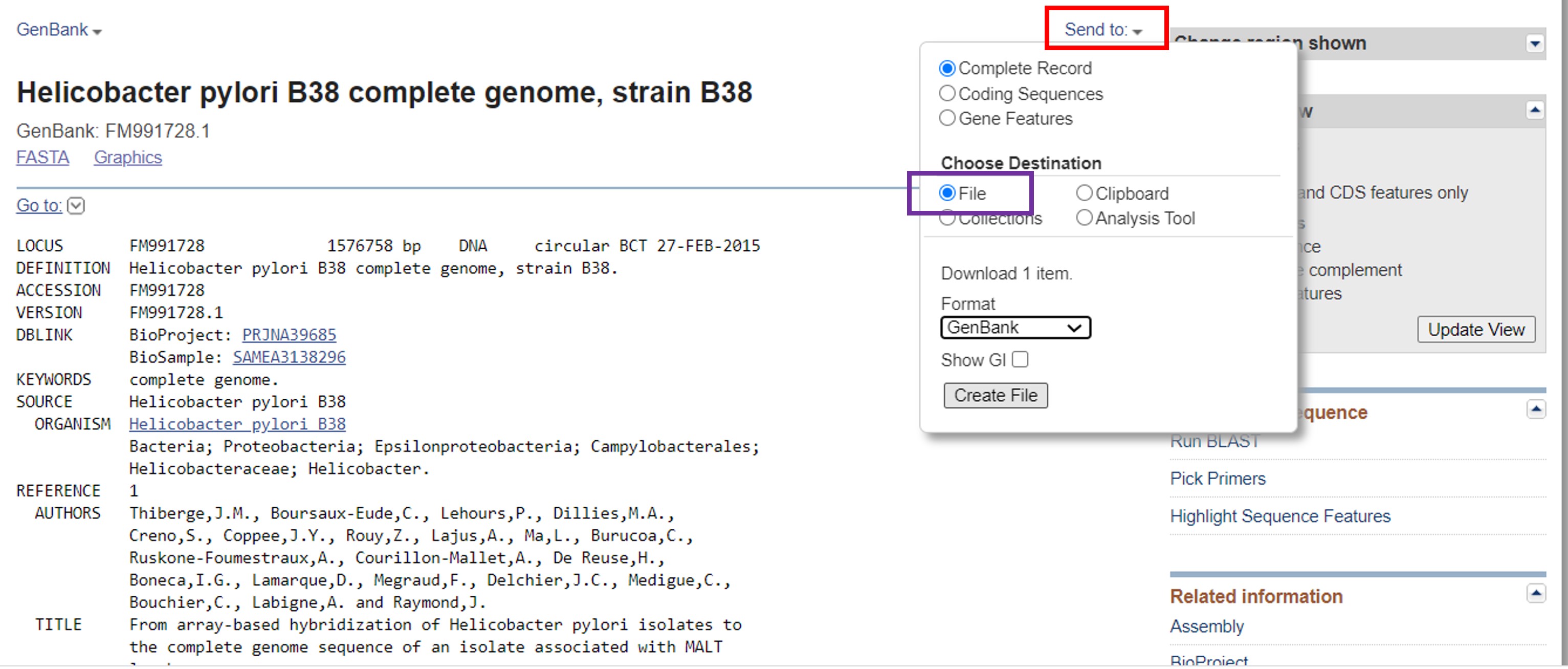
Figure 48: Bacterial genomes gene information download
In assignment_7 we learned that the pbpa gene plays a role in the “Biosynthesis of murein sacculus and peptidoglycan”. The peptidoglycan (murein) sacculus is a unique and essential structural element in the cell wall of most bacteria. Enzymes that make this structure are targets of many antimicrobials. However, microbes are becoming more resistant to this class of antimicrobials. If we want to know all the genes that play a role in the “Biosynthesis of murein sacculus and peptidoglycan” we can search the genbank file using the bash command grep. This command searches for patterns and print the line(s) where the pattern is present.
- Inspect the genbank data file with the
lesscommand
Line 45 until 48 contain information on the gene. Line 48 starts with /function followed by a description. Thus, we need to search for lines starting with /function followed by the first few words of our search pattern. It would also be better to print the 3 to 4 lines before the /function line because those lines contain additional information such as the gene name:
grep -iE -B4 "/function.*Biosynthesis of murein sacculus" lesson_4_opdracht8_Hpylori_sequence.gb - Argument -i means case insensitive
- Argument -E means a special version of grep to use special characters to search for patterns
- Argument -B4 means print 4 extra lines which are before the line containing the pattern
(
grepalso has a -A argument meaning print lines after the line containing the pattern)
The pattern starts with /function followed by a dot and an asteriks (.*). The dot means any character and the asterisk means zero or more time. Thus the combination .* means actually zero or any combination of characters.
We have now selected all genes with a function description of “Biosynthesis of murein sacculus” and 4 extra lines of information (Figure 49)
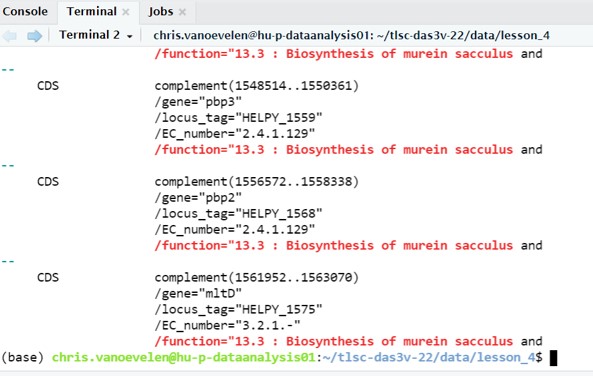
Figure 49: Bash grep command output
If we subsequently want to select the gene names we use the pipe symbol and searches for lines containing the characters /gene=
grep -iE -B4 "/function.*Biosynthesis of murein sacculus" lesson_4_opdracht8_Hpylori_sequence.gb | grep -iE "/gene="We are now left with a list of genes that are responsible for the “Biosynthesis of murein sacculus and peptidoglycan” (Figure 50). These genes serve as targets in de development for new antimicrobials
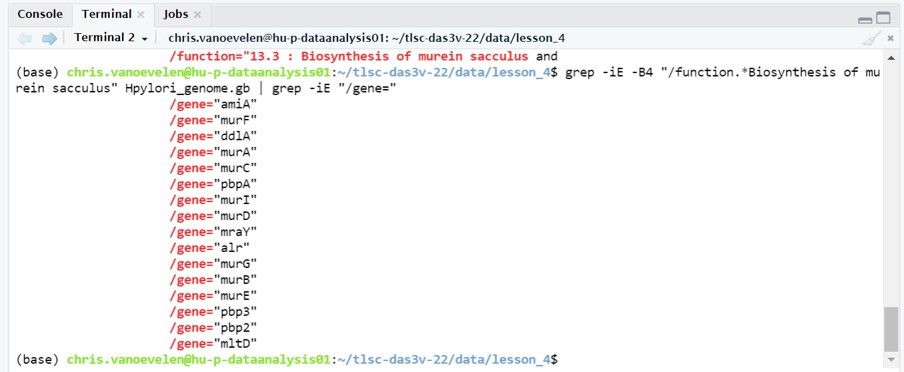
Figure 50: Bash grep command output
lesson_04_assignment
- Use grep to search for genes that are involved in “Biosynthesis of surface polysaccharides”
- How many genes are involved in “Biosynthesis of surface polysaccharides”
HINT: use the wc command to count lines of the output of the grep commands using a pipeline
- How many gene families are present in this list (look for similar gene names)
HINT: use the sort command to sort the output of the grep commands using a pipeline