Lesson_07 - Proteins
Learning objectives
The student is able:
- To explain the characteristics of a protein
- To translate a nucleotide sequence into an amino acid sequence
- To search for protein information using public databases (NCBI and uniprot)
- To use bash for the analysis of amino acid sequences
- To use NCBI blast to align amino acid sequences
Introduction
In the previous lessons we became acquainted with the structure of the genome, genes, DNA mutations and gene expression analysis. In this lesson we will focus on different features of proteins.
Interesting descriptive research questions are:
- What is the amino acid sequence?
- Which functional modules are present in the protein?
- Is the protein evolutionary conserved?
- Orthologs (between species)
- Paralogs (within the species)
- What is the function of the protein?
- What is the 3D structure of the protein?
- Is the protein modified by post-translational modifications (PTM) and what is the effect of the PTM on protein function?
- phosphorylation
- ubiquitination
- acetylation
- methylation
- glycosylation
- ..and many more
- Is the protein part of a protein-complex?
- What is the stability of the protein (half-life time of the protein)
- Does the protein require cleavage by proteinases for activation?
- Does the protein require co-factors for activation?
- Where is the protein located in the cell?
- nucleus
- cytosol
- membrane
- organelles
- DNA
- extra-cellular
- In which type of cell is the protein present?
To answer the above questions we can use different protein databases:
Translation
If we want to know the corresponding amino acid sequence we have to translate the nucleotide sequence into an amino acid sequence. In the NCBI database we can directly obtain the amino acid sequence of any given gene. However, if you acquire nucleotide sequences by sequencing experiments and want to know whether this sequence encode for a protein coding gene or quickly wants to scan for mutations at the amino acid level you have to translate the nucleotide sequence to an amino acid sequence yourself. We know that each (eukaryotic) coding sequence start with an ATG (= methionine) and ends with a stop codon (TAA, TAG, TGA). In between are the (minimal amounts of) triplets coding for amino acids.
There are several tools to translate a nucleotide sequence to an amino acid sequence. Here, we use the translate tool from the Expasy portal of bioinformatics:
In lesson 4 assignment.7 we obtained the nucleotide sequence of the human Smad2 gene:
- Copy file lesson_4_opdracht6_NM_005901.fa from lesson_04 to lesson_08 within your home directory
- Go to the Expasy webportal
- Search for the translate tool (use ctr f or Mac equivalent and search for “translate”)
- Click on “Browse the resource website”
- Paste the sequence of file lesson_4_opdracht6_NM_005901.fa into the window and use the default settings
- Press TRANSLATE!
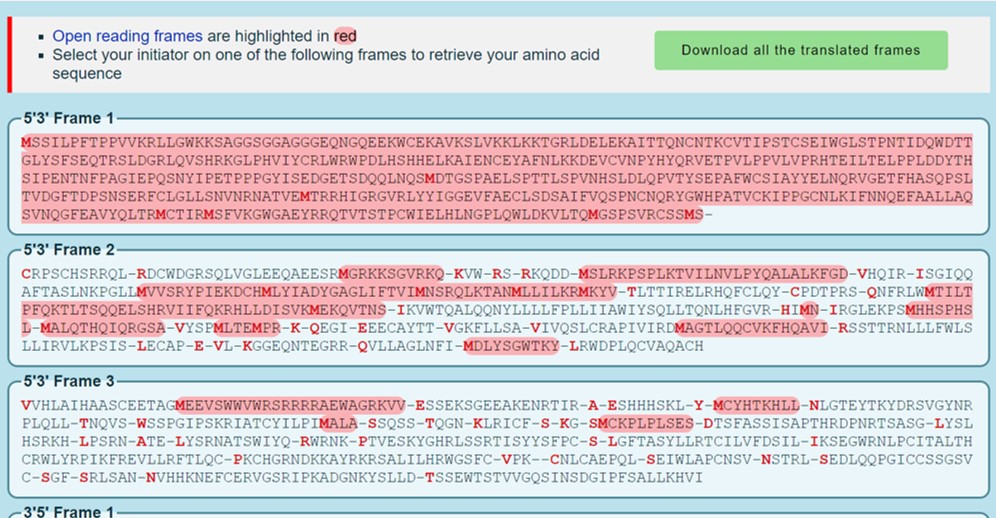
The Expasy translate tool prints out 6 reading frame (Figure 66)
lesson_07_assignment
Why are there 6 reading frames in the Expasy output?
When we use the Translate tool from Expasy we have to copy and paste the amino acids to a new file and save it. Alternatively we can write a bash script to automate this process:
- Copy bash script lesson_07_dna2protein.sh from the shared data folder to your home directory/lesson_07
- Check how the script is executed with the -h option
- Run the script with input file lesson_4_opdracht6_NM_005901.fa
- A new file is generated in your directory starting with protein_ followed by the name of the input file
Primary amino acid sequence
The primary amino acid sequences dictates protein folding and thus function. An important feature of the amino acid sequence is the frequency of non-polar and polar amino acids and in particular the organisation of these amino acids in blocks. How many blocks of non-polar and polar amino acids are present and what is the length of each block. In an aqueous environment, non-polar amino acid fold inwards into the protein structure and the polar amino acids outwards to form hydrogen bonds with the water. If a protein resides in the membrane, the non-polar amino acids will be on the surface of the protein structure to interact with the non-polar cell membrane. Most membrane bound receptors have a extra-cellular hydrophilic part which receives the ligand, a hydrophobic part that is associated with the membrane and a hydrophilic part present in the cytoplasm to relay the signal to other proteins such as the Smad2 protein
There are types of amino acids:
lesson_07_assignment
There are 20 different amino acids categorized in 4 groups:
- What are the 4 categories of amino acids?
- Which amino acids are in each group?
- Calculate the percentage amino acids for each group ?
NCBI protein database
To obtain information on a particular protein, we can use the NCBI protein database. In the previous lessons we have studied the molecular pathways of colorectal and gastric cancer and observed that the human Smad2/3 complex acts as an tumorsuppressor complex in colorectal cancer. When the Smad2/3 pathway is active it inhibits cell growth and thus proliferation. If mutations are present in the TGF\(\beta\), or the Smad2/3 genes and these mutations effect protein function, this inhibition is lost and cells can proliferate.
- Go to the NCBI protein database
- Search for human Smad2 protein (NP_001003652)
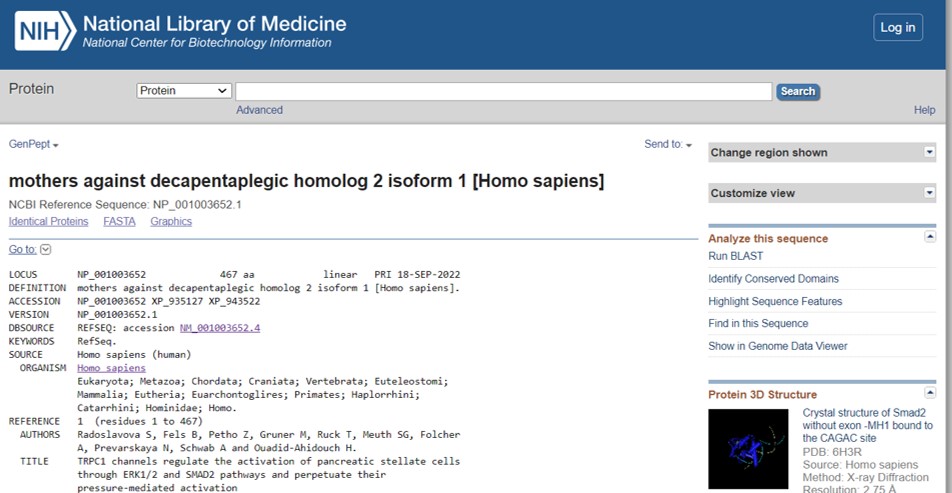
- Inspect the Smad2 protein information page (Figure 67)
We see some general information such as the protein ID and the length of the protein (467 amino acids). If you want to obtain the amino acid sequence in FASTA click on the FASTA link.
- Scroll down to view the feature section
This section lists functional domains and sites of post translational modification (PTM) such as phosphorylation and acetylation. These chemical groups are covalently attached to the protein and effect protein function. A more graphical view of all protein features can be viewed by clicking on the Graphics link on top of the information page (adjacent to the FASTA link)
lesson_07_assignment
- Which three amino acids can generally be phosphorylated in a protein sequence?
- How many amino acids become phosphorylated in Smad2?
- How many amino acids become acetylated in Smad2?
On the right hand side of the protein information page are additional links to analyze the protein sequence (Figure 67:
- Run BLAST
- Identify conserved domains
NCBI protein blast
BLAST analysis is used to:
- Identify mutations in biological sequences when compared to a wild-type sequence
- Identify unknown sequences:
- From which species?
- From which region of the genome?
- Identify paralogous sequences (within a species)
- Identify orthologous sequences (between species, showing a evolutionary relationship)
Here, we will use BLAST to identify paralogous sequences.
IMPORTANT: BLAST has different versions depending on whether the input sequence consists of nucleotides or amino acids. Be sure to select the right version for your analysis
To identify proteins which are similar to Smad2 at the amino acid level we will use protein BLAST. Proteins with a similar sequence could have similar functions and possibly work together in the same pathway. From the KEGG pathway analysis we already know that the Smad protein family consist of Smad1,2,3,4,5,6,7,9. Using BLAST we can identify at the amino acid levels where those proteins are most similar:
- Within the NCBI protein Smad2 information page click on Run BLAST
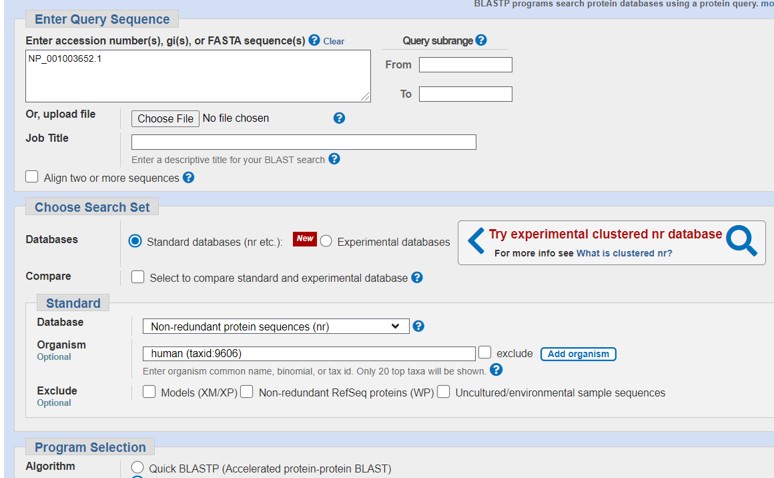
- Fill out the parameters as shown in Figure 68:
- Fill out human (taxid:9606) in the ORGANISM section to restrict the search for human only
- Select database “RefSeq Select proteins (ref_seq_select)”
- Scroll down, check “show results in a new window” and click on BLAST
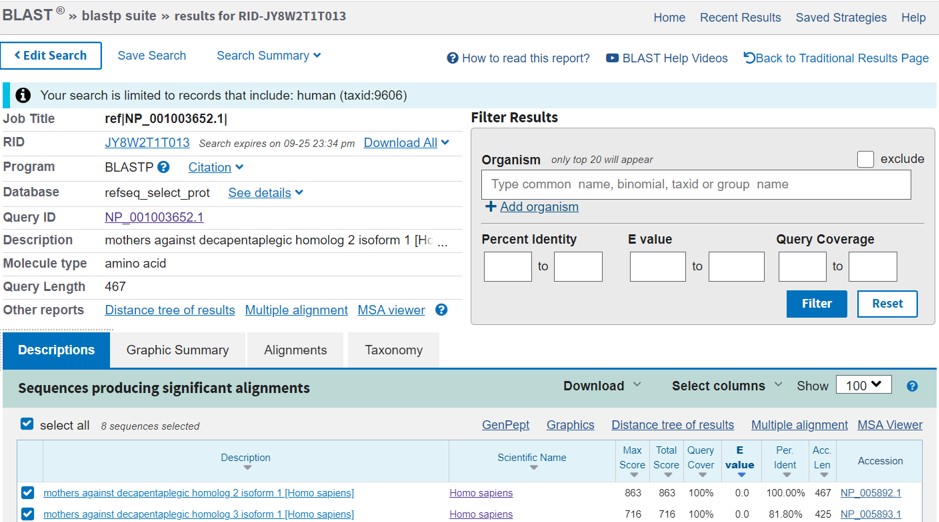
The output of the protein BLAST analysis consist of several panels (Figure 69)
lesson_07_assignment
- Which proteins shows highest / lowest similarity with the Smad2 protein (exclusing Smad2 itself)?
- Which part of the Smad7 sequence (position) shows similarity to the Smad2 protein?
protein domains
The protein paradigm is to predict the function of a protein based on the primary amino acid sequence. An important aspect of this question is to identify similar regions which are evolutionary conserved. These evolutionary conserved regions often fold into stand-alone structures with a specific molecular task. These regions are categorized as functional protein domains and can be considered as the building block of proteins. These domains are “reused” in proteins and the combination of domains defines the function of the protein. Proteins with similar domains belong to the same protein families, for example families of transcription factors, membrane-bound receptors, enzymes or kinases. These domains are most often coded by one exon and therefore mRNA splicing results in the deletion of functional domains and thus a protein with a different function.
It is also important to note that the effect of a mutation in the coding sequence of gene depends on where the mutation is located within the gene; which domain is affected. Secondly, the effect of an amino acid replacement depends on whether the new amino acid is from the same category (= same properties) or from a different category. If an polar amino acid is replaced by another polar amino acid then presumably this will have a less dramatic effect compared to a replacement by an amino acid from another category.
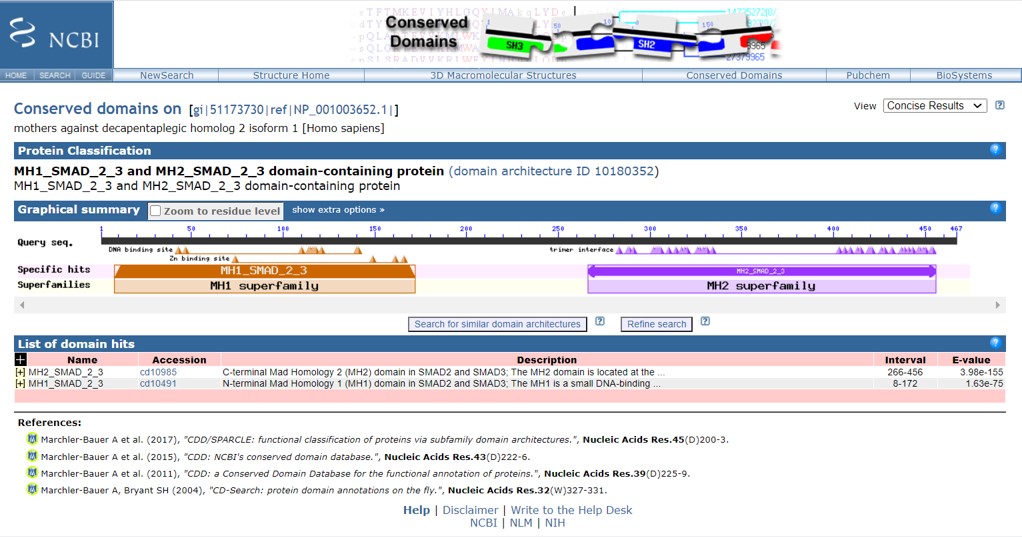
- Within the human Smad2 protein information page click “Identify Conserved Domains” (Figure 70)
The human Smad2 protein contains two conserved domains: MH1 and MH2. To obtain information on the function of the MH1 and MH2 domains, point with the cursor to the name of the domain (an information panel will appear) or go to the “List of domains hits” and click on the + sign to obtain information
lesson_07_assignment
- What are the positions of domains MH1 and MH2 in the human Smad2 protein?
- What is the function of the MH1 domain?
- What is the function of the MH2 domain?
Uniprot database
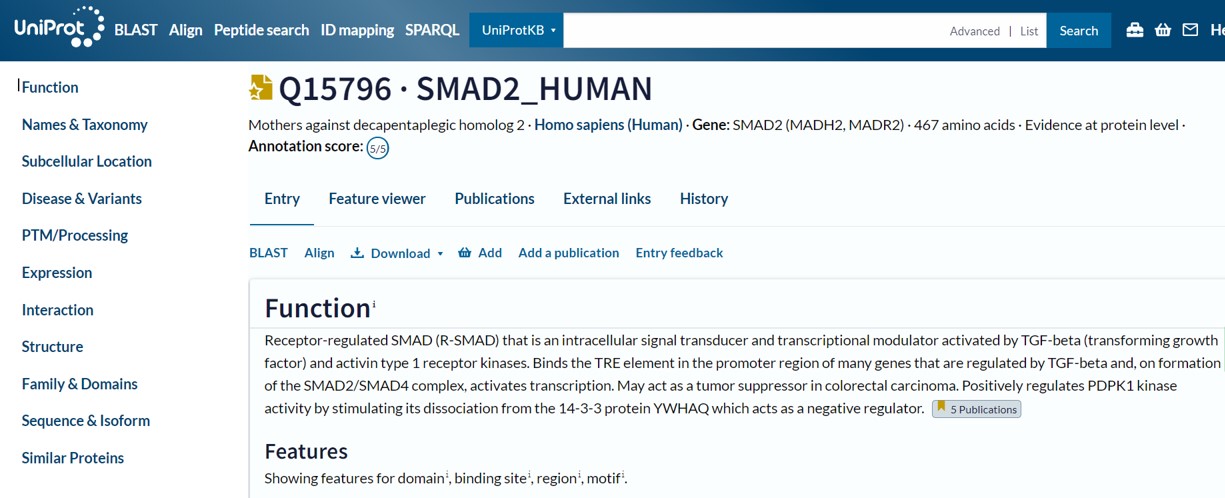
Another comprehensive protein database is uniprot. This database provides similar information as the NCBI protein database and numerous interactive tools to study your protein of interest:
- Search for human Smad2 protein
The navigation bar on on the left hand side contains links to the different sections containing protein information. A more graphical view of protein features can be accessed using the “Feature viewer” link (Figure 71)
The information page starts with the “Function” section. This section has several subsection:
- Features
- GO annotations (= Gene ontology -> description of location, function and molecular processes)
- Keywords
- Enzyme and pathway databases
The “Function” section provides general information on your protein of interest. To obtain more specific information go to the other subsection listed in the navigation bar on the left hand side
lesson_07_assignment
- Go to the “PTM/processing” section
- Which amino acids of human Smad2 protein are phosphorylated by TGFBR1
- Go to the “Disease & Variants” section
Mutations in the Smad2 protein can also cause (congenital heart disease)[https://medlineplus.gov/congenitalheartdefects.html]
- Describe the positions of the mutations that associated with this disease and the change in amino acids
- Go to the “Sequence & Isoforms” section
- How many isoforms exist of the human Smad2 protein
- What is the Mw (in KDa) of isoform 1.
Use the “Tools” option within the amino acid sequence
- Go to the “Interaction” section
This section provides a list of proteins which have been experimentally demonstrated to interact with Smad2. In lesson_3_assignment 3C we learned that SARA interacts with the Smad2 protein. However, this gene is not in the list. The reason is that the SARA protein has been renamed (and indeed the new name is included in the Interaction list)
- What is the official name of the human SARA protein?
HINT: Use one of the NCBI databases to obtain the original name
Bash: protein analysis
In addition to the protein databases we can use several bash functions to analyse protein sequences. For example, to count how many serine amino acids are present in the Smad2 protein we can make use of the bash grep command. (A summary file of the one letter abbreviations of the twenty amino acids and their categories is present in the general data folder lesson_07).
The first line of a fasta sequence contains a description of the sequence and not the sequence itself. Therefore we have to skip the first line for our analysis using the tail command and use the output of the tail command as input for the grep command:
tail -n+2 protein_lesson_4_opdracht6_NM_005901.fa | grep -i "s"
The tail argument -n+2 means print from line 2 until the end.
By default grep prints the complete line of the input file where a match is found. This way we can’t count the number of serines. To only print the match itself we use the -o argument. In combination with the wc -l command we can now count how many matches are present:
tail -n+2 protein_lesson_4_opdracht6_NM_005901.fa | grep -io "s" | wc -l
If we want to count the number of non-polar amino acids we can count them one by one by repeating the above pipeline. But we can also use special feature of grep. To activate the special features of grep we have to use the -E argument. To search for all non-polar amino acids we provide the letters of the non-polar amino acids to a so-called character class:
tail -n+2 protein_lesson_4_opdracht6_NM_005901.fa | grep -ioE "[AGILMFPWV]" | wc -l
The character class [AGILMFPWV] means that grep searches for any single characters which are listed between the [ ].
The output of the script shows 210 non-polar amino acids.
lesson_07_assignment
- How many polar, acidic and basic amino acids are present in the Smad2 sequence?
- How many Serines, Threonines and Tyrosines are present in the Smad2 protein sequence?
With grep it is also possible to search for multiple patterns. The PPPGY pattern is involved in protein-protein interactions and the SSMS pattern is a phosphorylation site. To search for both patterns we make use of the alternating symbol which is the | sign.
NOTE: the | sign in bash context is used to create pipelines. In grep context it means alternating: search for pattern1 or pattern2
In the following examples the matches are shown in the sequence itself and not counted. If you want to count the number of matches use the grep -o argument and the wc -l command as shown in the example above:
tail -n+2 protein_lesson_4_opdracht6_NM_005901.fa | grep -iE "PPPGY|SSMS"
With grep it is also possible to search for complex patterns. For example if we are searching for a pattern that start with PP and end with LL and in between there are four amino acids of any type:
tail -n+2 protein_lesson_4_opdracht6_NM_005901.fa | grep -iE "PP....LL"
A dot in a grep search pattern means any character. Thus “PP….LL” means start with PP followed by any of 4 characters and ends with LL.
But what if we want to search for a pattern that starts with PP and ends with E and in between there could be 1 to 10 amino acids of any type:
tail -n+2 protein_lesson_4_opdracht6_NM_005901.fa | grep -ioE "PP.{1,10}E"
Here, we see that the dot is followed by {1,10}. This is a so called quantifier meaning that the dot (= any character) can be present in the pattern for 1 up to a maximum of 10 times.
lesson_07_assignment
How many patterns are present in the human Smad2 protein that starts with E and ends with E and in between there could be 1 to 5 amino acids of any type?
How many patterns are present in the human Smad2 protein with the previous pattern and an additional pattern that starts with a P and in between any of three amino acids and ends with a D ? Write one pipeline.
A researcher wrote a bash script to easily scan the amino acid composition of a protein sequence. Script lesson_07_AA.sh is located in the shared folder.
- Copy this script to your home directory lesson_07
To use this script we have to know how to execute it. The writer of (any) script has to provide information on how to execute the script. This information is generally provided by the -h argument:
Use this script to answer to following questions:
lesson_07_assignment
What are the percentage of acidic, basic, non-polar and polar amino acids of the human Smad2 protein sequence?
Compare these percentage to the expected percentages (assignment_02). What is your conclusion?
Obtain the amino acid sequence of human rhodopsin (NP_000530). Save as lesson_07_opdracht1_NP_000530.fa on the server
In which compartment of the cell is Rhodopsin located?
HINT: use the NCBI gene information page or uniprot database
What is the percentage of acidic, basic, non-polar and polar amino acids of the human Rhodopsin sequence?
Compare the values to the expected values and to the values of the human Smad2 sequence. What is your conclusion?
Using the script we can also count the presence of individual amino acids or detect amino acid motifs. A motif is a short stretch of amino acids that is evolutionary conserved and thus plays an important role in protein function. When a pattern is found the script will also print the position within the sequence. Use the -h option to read how to use the script to found patterns
lesson_07_assignment
At what position in the human Rhodopsin protein is the NPxxY present?
(x represent any amino acid)