Les8 - Parametrische testen(2)
Leerdoelen
Uitvoeren van parametrische toetsen:
- Ongepaarde ANOVA
- Gepaarde ANOVA
- Two-way ANOVA
- Non-parametrische toetsen
Introductie
Om een ANOVA test te doen moet de experimentele opzet en de data aan een aantal voorwaarden voldoen:
- Je wilt een verschil aantonen tussen de controle groep en experimentele groep(en) (een vergelijkende onderzoeksvraag)
- De meetwaarden zijn kwantitatief
- Geen uitbijters in de dataset
- De data is normaal verdeeld (dit wordt voor ANOVA anders bepaald dan voor de t-testen)
- De varianties van de experimentele groepen zijn gelijk
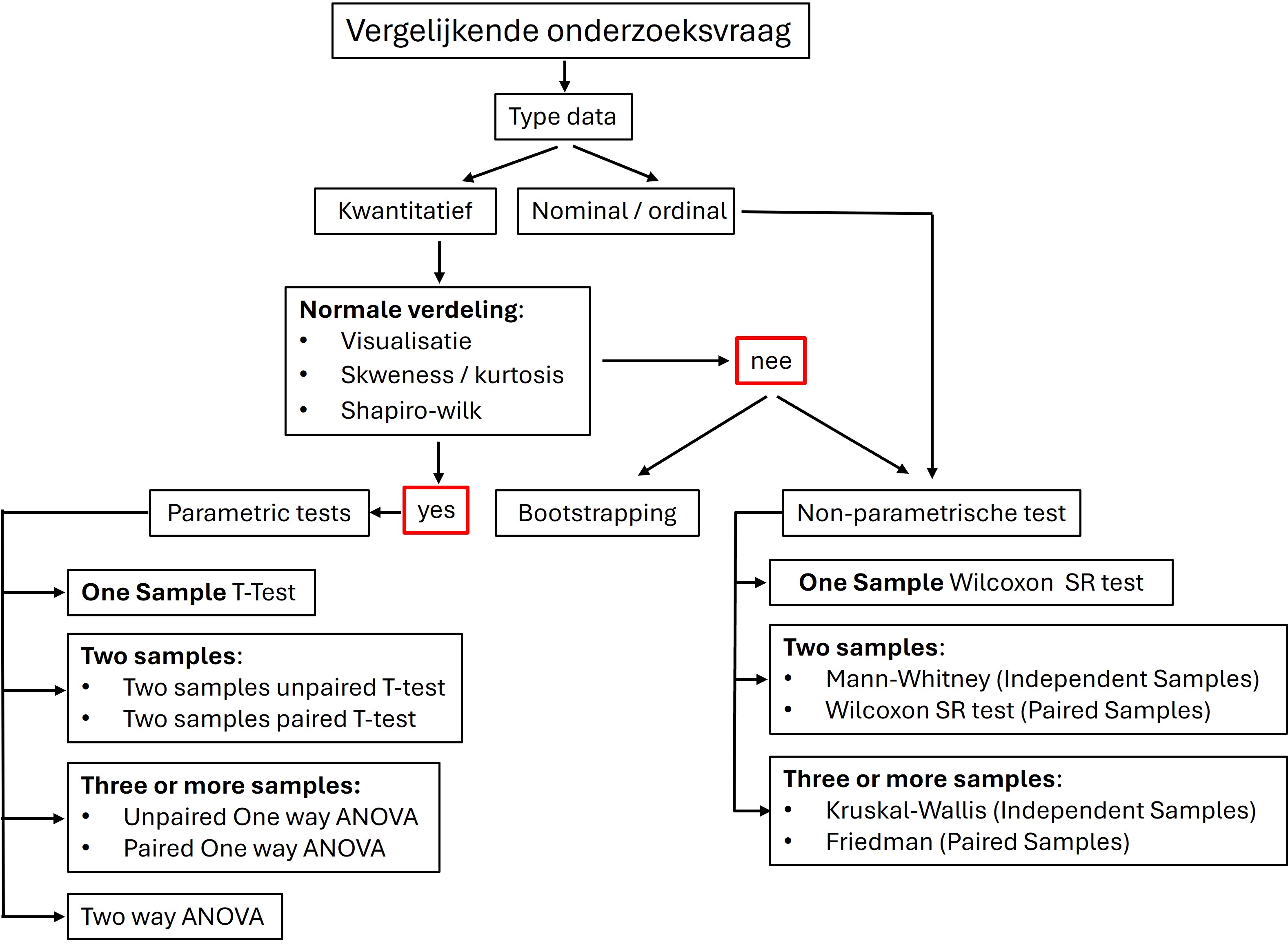
Figure 59: Beslissingsboom statistische testen
In de vorige les is de t-test behandeld voor twee steekproeven. In deze les leggen we de ANOVA uit voor drie of meer steekproeven en de two-way ANOVA voor een multiple factor ontwerp.
ANOVA is een afkorting voor Analysis Of Variance en is in feite weer een signaal-ruis verhouding zoals we eerder bij de t-test hebben gezien.
Unpaired one way ANOVA
Gebruik de “Unpaired one way ANOVA” bij een ongepaard OFAT experiment waarbij er drie of meer onafhankelijke groepen zijn, die ieder één keer zijn gemeten (zie figuur 60, linker figuur).
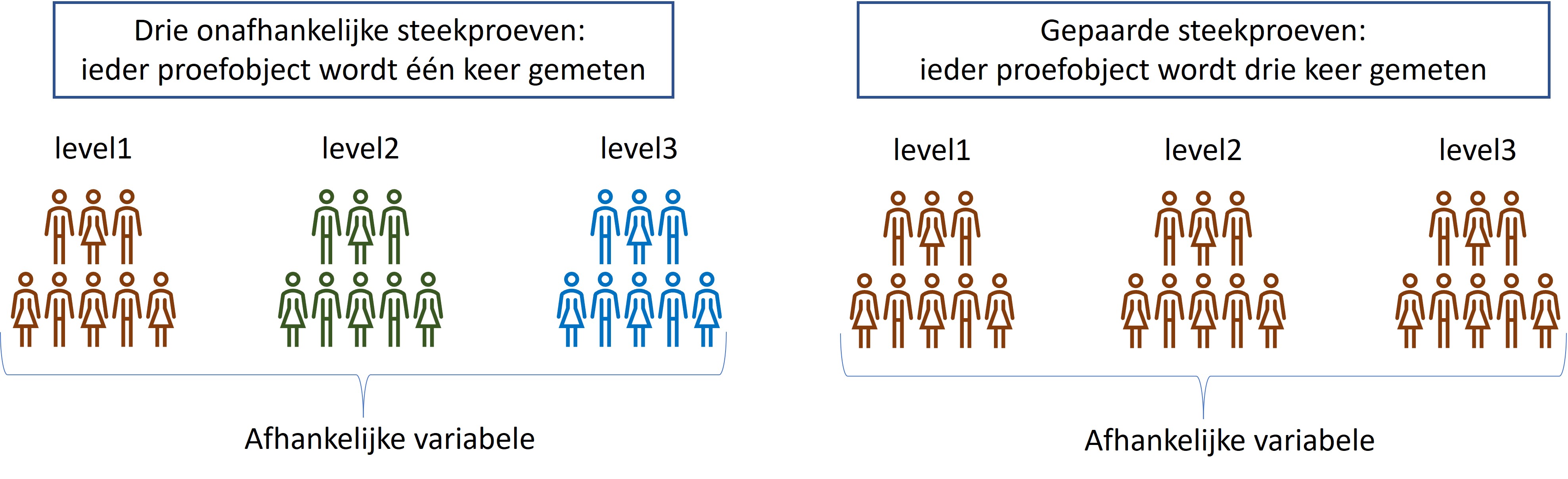
Figure 60: Overzicht van ongepaard en gepaard OFAT experiment met drie groepen.
De F-statistiek van een ongepaarde ANOVA test is:
\[ F = \frac{Variantie\;tussen\;de\;groepen}{Variantie\;binnen\;de\;groepen}= \frac{MS_{between}}{MS_{within}} \] De “Variantie tussen de groepen” is het signaal van het experiment (teller van de formule). Stel we hebben drie onafhankelijk experimentele groepen met ieder 5 meetpunten (zie figuur 61)
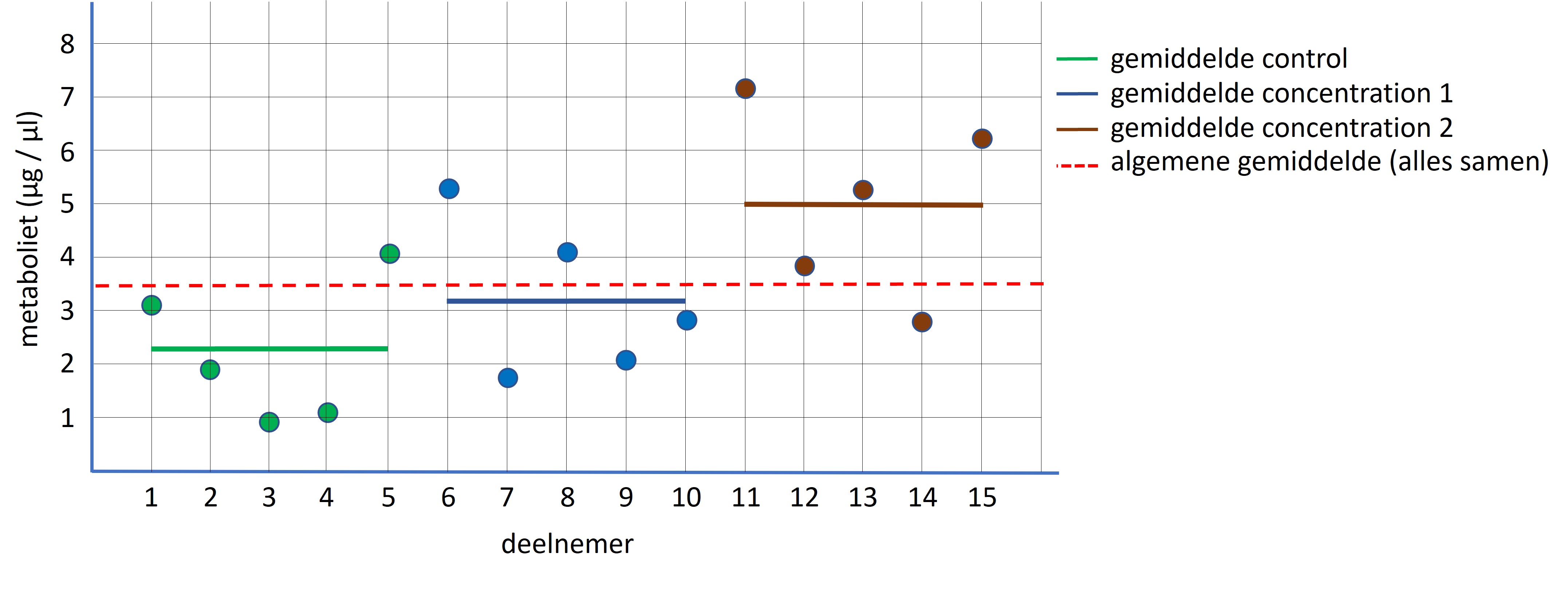
Figure 61: Ongepaarde ANOVA: algemene en steekproef gemiddelden
De drie steekproeven komen initiëel uit dezelfde populatie en als er geen effect is van de experimnentele behandeling dan zijn de gemiddelden van de steekproeven min of meer gelijk en dus ook gelijk aan het algemene gemiddelden (zie figuur 62, rode stippellijn; gemiddelde gebaseerd op alle datapunten). Hoe meer de groepen onderling van elkaar verschillen (de behandeling heeft een bepaald effect) hoe groter het verschil tussen het gemiddelde van iedere groep en het algemene gemiddelde. Dit verschil wordt uitgedrukt met de variantie waarbij eerst voor iedere steekproef het verschil wordt berekend tussen het gemiddelde van iedere steekproef en het algemene gemiddelde (zie figuur 62 verschil tussen de gekleurde verticale pijlen en de rode stippellijn). Dit verschil wordt gekwadradeerd en vermenigvuldigd met de grootte van de steekproef (n) en van alle groepen bij elkaar opgeteld. We delen deze waarde ook door het aantal vrijheidsgraden, wat overeenkomt met (het aantal groepen -1) = 3 -1 = 2. Deze waarde is dus het signaal van het experiment en hoe groter het signaal, hoe groter de kans dat de experimentele behandeling een effect heeft gehad.
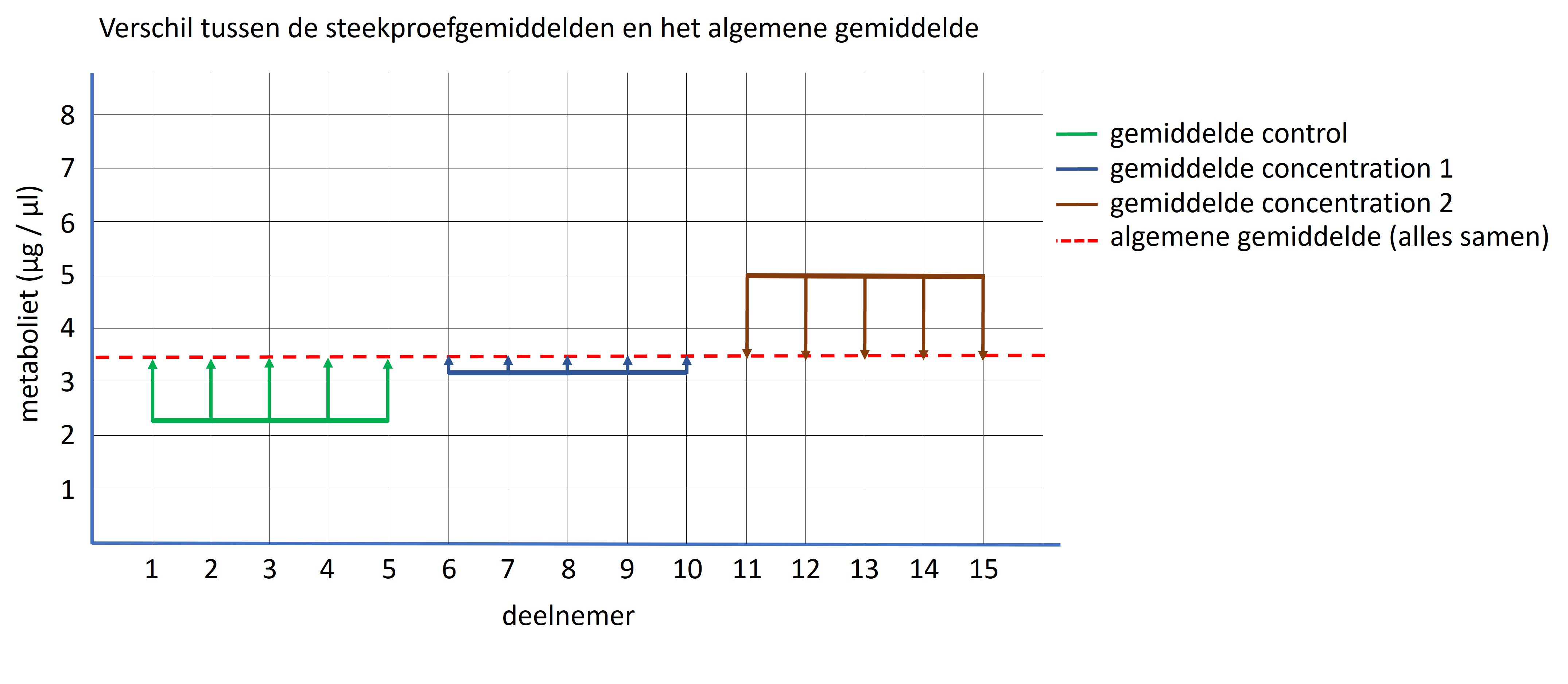
Figure 62: Verschil tussen algemene gemiddelde en steekproef gemiddelden
De tweede stap is het berekenen van de “variantie binnen de groepen” (noemer van de formule) en dit is de ruis van het experiment. De ruis wordt uitgedrukt door het verschil van ieder datapunt ten opzichte het steekproefgemiddelde uit te rekenen. Deze verschillen worden ook wel de residuals genoemd en worden verder gebruikt om te bepalen of de data normaal is verdeeld. Deze verschillen worden gekwadradeerd en ook bij elkaar opgeteld om het totale verschil binnen de steekproeven te berekenen (= het verschil dat we uitsluitend op basis van toeval verwachten). We delen deze waarde ook door de vrijheidsgraad, wat het totale aantal metingen is -1 = (3 * 5) -1 = 15 -1 = 14.
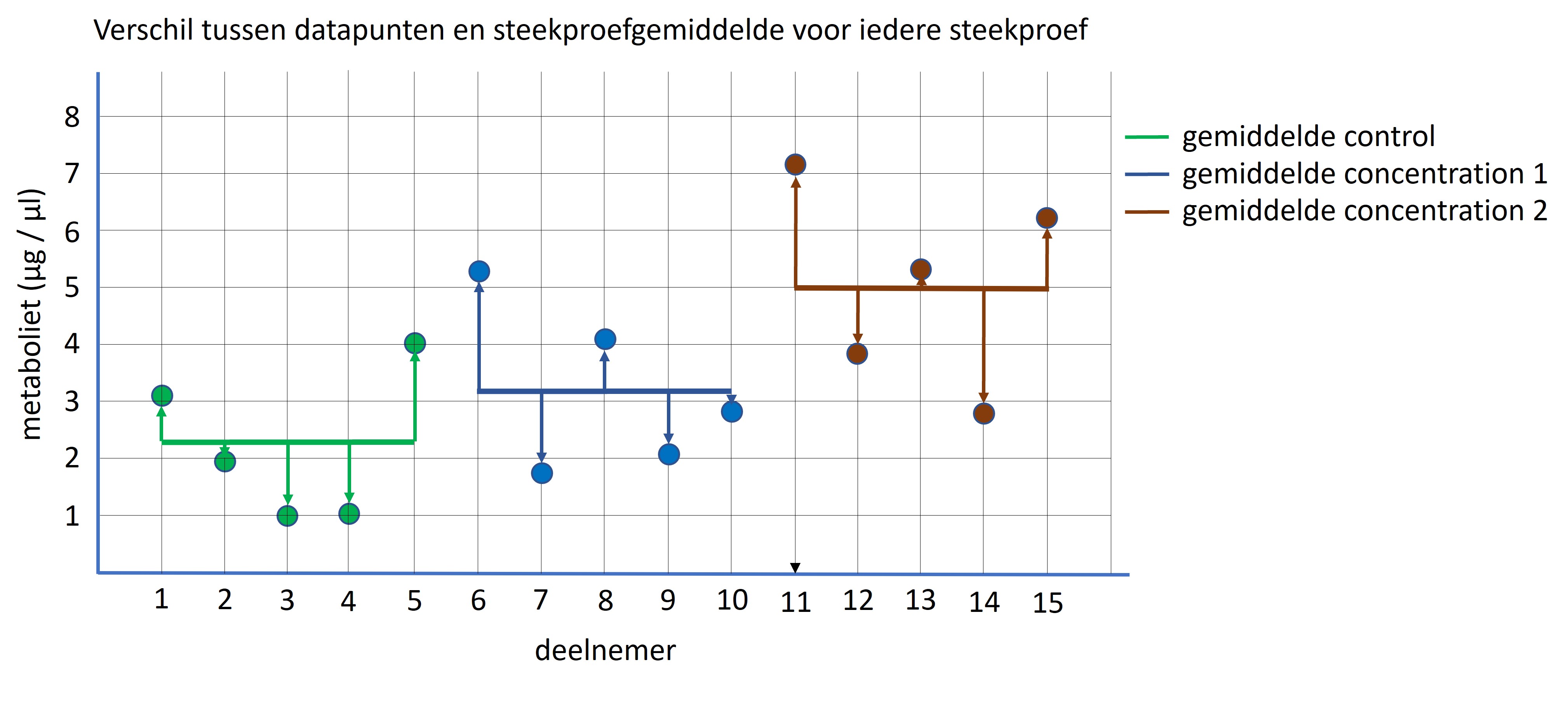
Figure 63: Verschil binnen de steekproeven. Dit is een maat voor ruis van het experiment
Vervolgens berekenen we de F-statistiek. De F-statistiek is verhouding tussen het signaal (verschillen tussen de steekproeven) en de ruis van het experiment (verschillen binnen de steekproeven). Vervolgens wordt m.b.v. de F-verdeling uitgerekend wat de kans is om een bepaalde F-waarde te meten onder de aanname dat de behandeling niet heeft gewerkt. Als een behandeling niet werkt, is het signaal min of meer gelijk aan de ruis van het experiment en de F waarde ligt rond de 1. Dit wordt formeel vastgelegd met de nul-hypothese (H0):
H0: \(\mu1\) = \(\mu2\) = \(\mu3\) (de gemiddelden van iedere groep zijn gelijk)
H1: De gemiddelden van de groepen zijn niet gelijk
(Deze hypothese stelt dat er ergens een verschil is tussen de groepen maar geeft niet aan tussen welke groepen!)
Intiutief voel je aan dat de kans steeds kleiner wordt om per toeval grote verschillen te meten tussen drie steekproeven uit dezelfde beginpopulatie. Als deze kans (p-waarde) kleiner is dan 0,05 dan verwerpen we de H0 en wordt de H1 geaccepteerd. Er is wel een verschil tussen de steekproefgemiddelden. Dit verschil berust niet op toeval.
P-waarde van de t-test ≥ 0,05: accepteer H0. Er is geen statistisch significant verschil tussen de populaties.
P-waarde van de t-test < 0,05: verwerp H0 en accepteer H1. Er is wel statistisch significant verschil tussen de populaties.
Voor het uitvoeren van een ANOVA test moeten we eerst bepalen of:
(1) De data (residuals) normaal verdeeld zijn
(2) de varianties tussen de groepen gelijk zijn met behulp van de Levene’s test zoals voor de ongepaarde t-test. (Als de p-waarde van de Levene’s test < 0,05 dan wordt de Welch ANOVA uitgevoerd die rekening houdt met de ongelijke varianties. Dit is een variant van de ANOVA. De p-waarde wordt groter en is dus conservatiever.)
We gebruiken de interne dataset PlantGrowth om een ongepaarde ANOVA uit te voeren. De PlantGrowth data is al tidy.
- Laad de juiste libraries voor de analyse
library(tidyverse)
library(ggplot2)
library(moments)
library(car)- Zet de kolom met de onafhankelijke variabele (=factor) om naar een factor variabele
# de kolom 'Species' omzetten naar een factor variabele
PlantGrowth$group <-as.factor(PlantGrowth$group)- Beschrijvende statistiek
# Bereken het gemiddelde, sd en het aantal metingen per experimentele groep
# Dit gebruiken we later om een staafdiagram te maken
plantgrowth_summarize <-PlantGrowth |>
group_by(group) |>
summarize(mean_weight=mean(weight),
sd_weight=sd(weight),
n_weight=length(weight))
# Aantal metingen is 10 per groep.
# Controleer voor normaal verdeling(4a) Test of de data normaal is verdeeld
Hiervoor gebruiken we niet de ruwe data (meetpunten per experimentele groep) maar de residuals (zie figuur 63). De eerste stap is het verkrijgen van de residuals:
# Verkrijg de residuals met behulp van de aov() functie
model_plantgrowth <- aov(data= PlantGrowth,
weight ~ group)
res_plantgrowth <- residuals(model_plantgrowth)
# maak een tibble aan met de residuals
res_plantgrowth_tibble <- tibble(res=res_plantgrowth)(4b) Test of de residuals normaal verdeeld zijn met Shapiro-Wilk test en qq-plot
# Test of de residuals normaal verdeeld zijn met de Shapiro-Wilk test
shapiro.test(res_plantgrowth_tibble$res)$p.value |> round(digits=2)
# Maak een qq-plot met de residuals
res_plantgrowth_tibble |>
ggplot(aes(sample = res)) +
stat_qq() +
stat_qq_line(color = "red") +
theme_minimal()
# Conclusie. P-waarde SW test is 0.44. Accepteer H~0~. De data is normaal verdeeld
# Dit wordt verder ondersteunt met de qq-plot....
# hoewel het einde van de punten niet op de referentielijn liggen. De meeste punten wel.(4c) Optioneeel (want data is al normaal verdeeld): Test of de data uitbijters bevat:
PlantGrowth |>
ggplot(aes(x=group, y=weight))+
geom_boxplot()
# conclusie: de data bevat uitbijters maar de data is evengoed normaal verdeeld
# De uitbijter verstoord niet de normaal verdeling van de data
# Dus we laten de uitbijters zitten- Visualiseer de data
# Staafdiagram + errorbars met behulp van ggplot
# Gebruik object plantgrowth_summarize van stap (3) beschrijvende statistiek
plantgrowth_summarize |> ggplot( aes(x = group, y = mean_weight)) +
geom_col() +
geom_errorbar(
aes(ymin = mean_weight - sd_weight,
ymax = mean_weight + sd_weight),
width = 0.2) +
labs(
x = "group",
y = "weight",
title = "dry weight of plants of control and two experimental condition") +
theme_minimal()- Test of de varianties tussen de groepen gelijk zijn met de Levene’s test
# Levene's test (want aantal metingen = 10)
leveneTest(data=PlantGrowth, # data bestand waarop de Levene test moet worden uitgevoerd
weight~group) # formule die de afhankelijke variabele (links) en de onafhankelijke variabele (rechts) aangeeft
# conclusie: P-waarde = 0.3412. Accepteer de H~0~. De varianties zijn gelijk- Voer een ongepaarde ANOVA test uit
# Gebruik de functie oneway.test() voor ANOVA analyse
oneway.test(data=PlantGrowth,
weight ~ group,
var.equal=TRUE) # Geef aan dat de variantie gelijk zijn
# conclusie: P-waarde = 0.01591. Verwerp H~0~ en accepteer H~1~. Er is wel een verschil tussen de groepen.
# We weten echter niet tussen welke groepen!! Dit wordt getest met een post-hoc test (zie tekst hieronder)les8_opdracht
Test of er een verschil is in “Sepal.Length” tussen de drie iris soorten van de (interne) “iris” dataset
Klik voor het antwoord
# (1) Laad de libraries. Die zijn al geladen dus geen verdere instructies
# (2) de kolom 'Species' omzetten naar een factor variabele
iris$Species <- as.factor(iris$Species)
# (3) Beschrijvende statistiek: gemiddelde en sd (voor het maken van een staafdiagram)
iris_summarize <-iris |>
group_by(Species) |>
summarize(mean_pl=mean(Sepal.Length),
sd_pl=sd(Sepal.Length),
n_pl=length(Sepal.Length))
# (4) Test of de data normaal is verdeeld
# Er zijn 50 metingen per iris soort (zie R-object iris_summarize van stap 3
# Dus er hoeft niet getest te worden of de data normaal verdeeld is
# (4c) Test of de data uitbijters bevat:
iris |>
ggplot(aes(x=Species, y=Sepal.Length))+
geom_boxplot()
# virginica heeft 1 uitbijter. Maar omdat n=50 en de boxplot rederlijkerwijs symmetrisch is laten we de uitbijter zitten
# Als een (extreme) uitbijter aanwezig is die een grote invloed heeft op het gemiddelde, sd en de verdeling ...
# dan verwijderen we de uitbijter. In alle andere gevallen laten we de uitbijter zitten
# (5) Visualiseer de data
iris_summarize |>
ggplot(aes(Species, mean_pl))+
geom_col()+
geom_errorbar(
aes(ymin = mean_pl - sd_pl,
ymax = mean_pl + sd_pl),
width = 0.2) +
labs(
x = "Iris soort",
y = "Lengte van Sepal (cm)",
title = "Lengte van Sepal van drie verschillende Iris soorten") +
theme_minimal()
# (6) Test of de varianties tussen de groepen gelijk zijn met de Levene's test
leveneTest(data= iris,
Sepal.Length ~ Species)
# conclusie: p-waarde = 0.002259. Verwerp H~0~ en accepteer H~1~: varianties zijn niet gelijk
# (7) Voer een ANOVA test uit
oneway.test(data=iris,
Sepal.Length ~ Species,
var.equal = FALSE) # varianties zijn niet gelijk. Welch ANOVA wordt uitgevoerd
# conclusie: p-waarde = p-value < 2.2e-16. Verwerp H~0~ en accepteer H~1~: gemiddelden zijn niet gelijkPost-hoc test: ongepaard
ANOVA test of er een verschil is tussen de groepen, maar de uitkomst geeft niet aan welke groepen verschillen. Maar wat is het nut van een ANOVA als we alleen kunnen zeggen dat er ergens een verschil is tussen de groepen? Kunnen we geen onafhankelijke Samples T-test uitvoeren tussen groep1/2, groep1/3 en groep2/3? Het antwoord is NEE (zie verklaring hieronder)
De nulhypothesetoetsing is een kansberekening en als de kans kleiner is dan 0,05 (= significantie niveau) om een bepaald verschil te meten tussen twee groepen dan noemen we het verschil statistisch significant. Dit verschil kan echter nog steeds door toeval worden verklaard. Dus in 1 van de 20 soortgelijke experimenten maken we een type I fout. We concluderen dat er een statistisch significant verschil is maar in werkelijkheid berust dit op toeval. Als we meerdere t-testen zouden doen op dezelfde data (zoals bij een ANOVA) dan maken dan vaker een type I fout. Dit wordt de “family-wise error rate” genoemd en dit kan op de volgende manier qworden berekend:
1 - (1 - significantie niveau)^aantal vergelijkingen^ x 100
1 test: 1 - (1 - 0,05)1 x 100 = 5%
2 tests: 1 - (1 - 0,05)2 x 100 = 9,75%
3 tests: 1 - (1 - 0,05)3 x 100 = 14,3%
Met drie vergelijkingen verhogen we dus de kans op een type I-fout van 5% naar 14,3%.
les8_opdracht
Een onderzoeker wil de genexpressieniveaus testen in levercellen die zijn blootgesteld aan PFAS. De onderzoeker heeft 20.000 genexpressiewaarden verkregen voor de controlegroep en de behandelde groep. De onderzoeker selecteert 10 genen en voert voor ieder gen een t-tests uit.
Bereken de “family-wise error rate”
Klik voor het antwoord
1 - (1 - 0,05)10 * 100 = 40,1 %Om voor deze “family-wise error rate” te corrigeren, kunnen we eerst een ANOVA uitvoeren om te controleren of er een verschil is tussen de groepen. Zo ja, dan gaan we verder met een post-hoc analyse. Deze analyse voert de paarsgewijze vergelijking tussen de groepen uit, maar corrigeert voor de toename in de type I-fout en houdt het totale foutpercentage op 5%. Er bestaan verschillende post-hoc-testen, waarvan de Bonferroni het meest gebruikt wordt.
R code om een post-hoc test uit te voeren:
pairwise.t.test(iris$Sepal.Length, # afhankelijke variabele
iris$Species, # onafhankelijke variabele
paired = FALSE,
p.adjust.method = "bonferroni") ##
## Pairwise comparisons using t tests with pooled SD
##
## data: iris$Sepal.Length and iris$Species
##
## setosa versicolor
## versicolor 2.6e-15 -
## virginica < 2e-16 8.3e-09
##
## P value adjustment method: bonferroniDe verschillende groepen staan weergegeven in een tabel met de p-waarden. In de iris dataset zien we dat alle p-waarden < 0,05. Er is een statistisch significant verschil in de gemiddelde Sepal.Length tussen:
versicolor en setosa (p-waarde = 2.6e-15)
versicolor en virginica (p-waarde = 8.3e-09)
virginica en setosa (p-waarde = 2e-16)
Paired one way ANOVA
Gebruik de “paired one-way ANOVA” bij een gepaard OFAT experiment waarbij één groep meerdere keren wordt gemeten (zie figuur 60, rechter figuur).
De F-statistiek van een gepaarde ANOVA test is:
\[ F = \frac{Variantie\;tussen\;de\;groepen}{Variantie\;binnen\;de\;groepen}= \frac{MS_{between}}{MS_{error}} \]
De “Variantie tussen de groepen” is het signaal van het experiment (teller van de formule). Een voorbeeld van een paired ANOVA (= repeated measurement ANOVA) is dat een groep patiënten een trainingsschema uitvoert om chronische ontstekingen te verminderen. De aanwezigheid van een eiwit in hun bloed, dat dient als marker voor ontsteking, wordt gemeten voor en na de verschillende tijdstippen van het trainingsschema (zie figuur 64):
meting1: vóór de start van de training
meting2: na 4 weken
meting3: na 8 weken
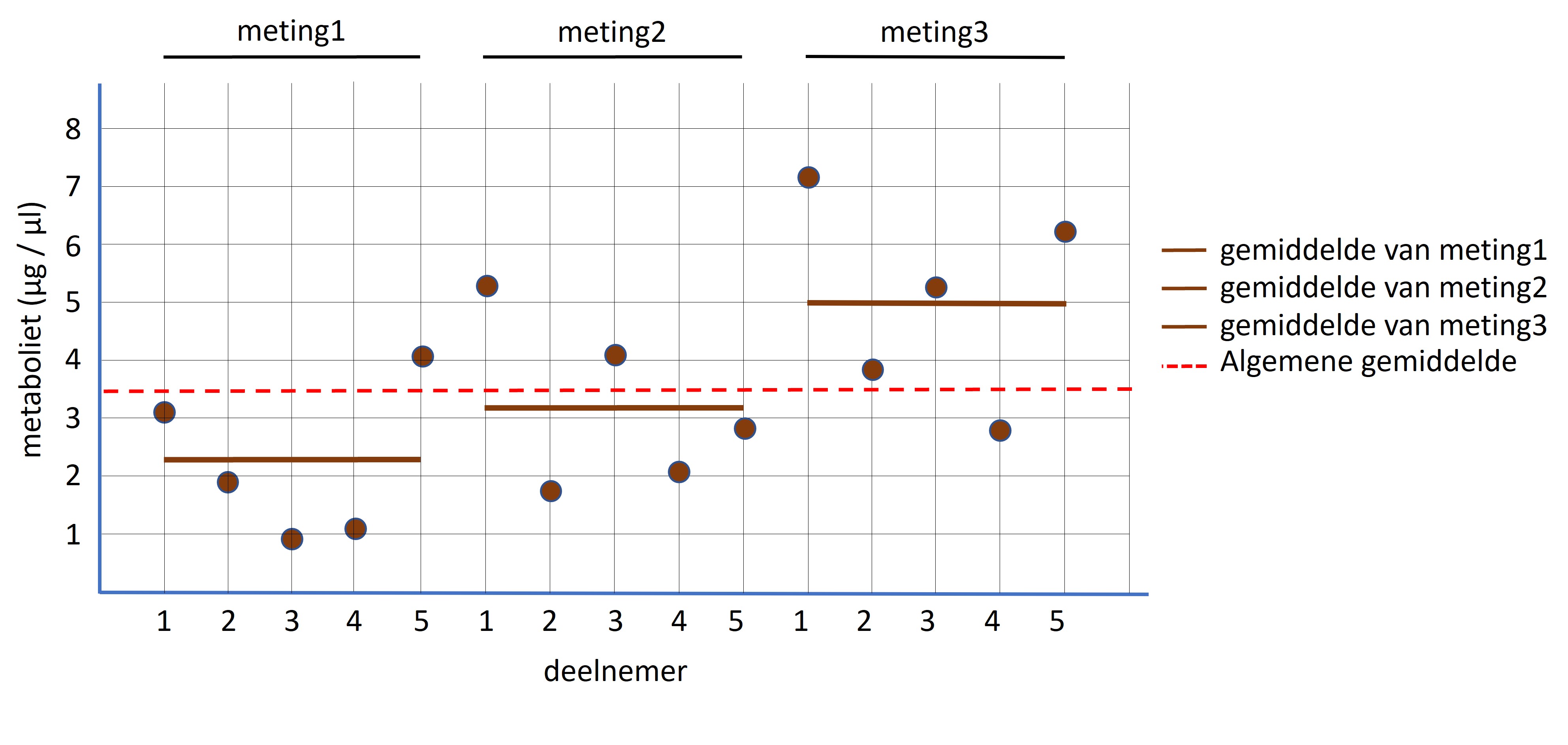
Figure 64: Gepaarde ANOVA: gemiddelde per meting en het algemene gemiddelde
De ANOVA-analyse voor gepaarde monsters volgt dezelfde logica zoals hierboven beschreven voor een ongepaarde ANOVA. Als de oefeningen geen effect hebben op de eiwitconcentratie dan zijn de gmiddelden van iedere meting min of meer gelijk en dus ook gelijk aan het algemene gemiddelde, zie figuur 65, rode stippellijn; gemiddelde gebaseerd op alle datapunten).
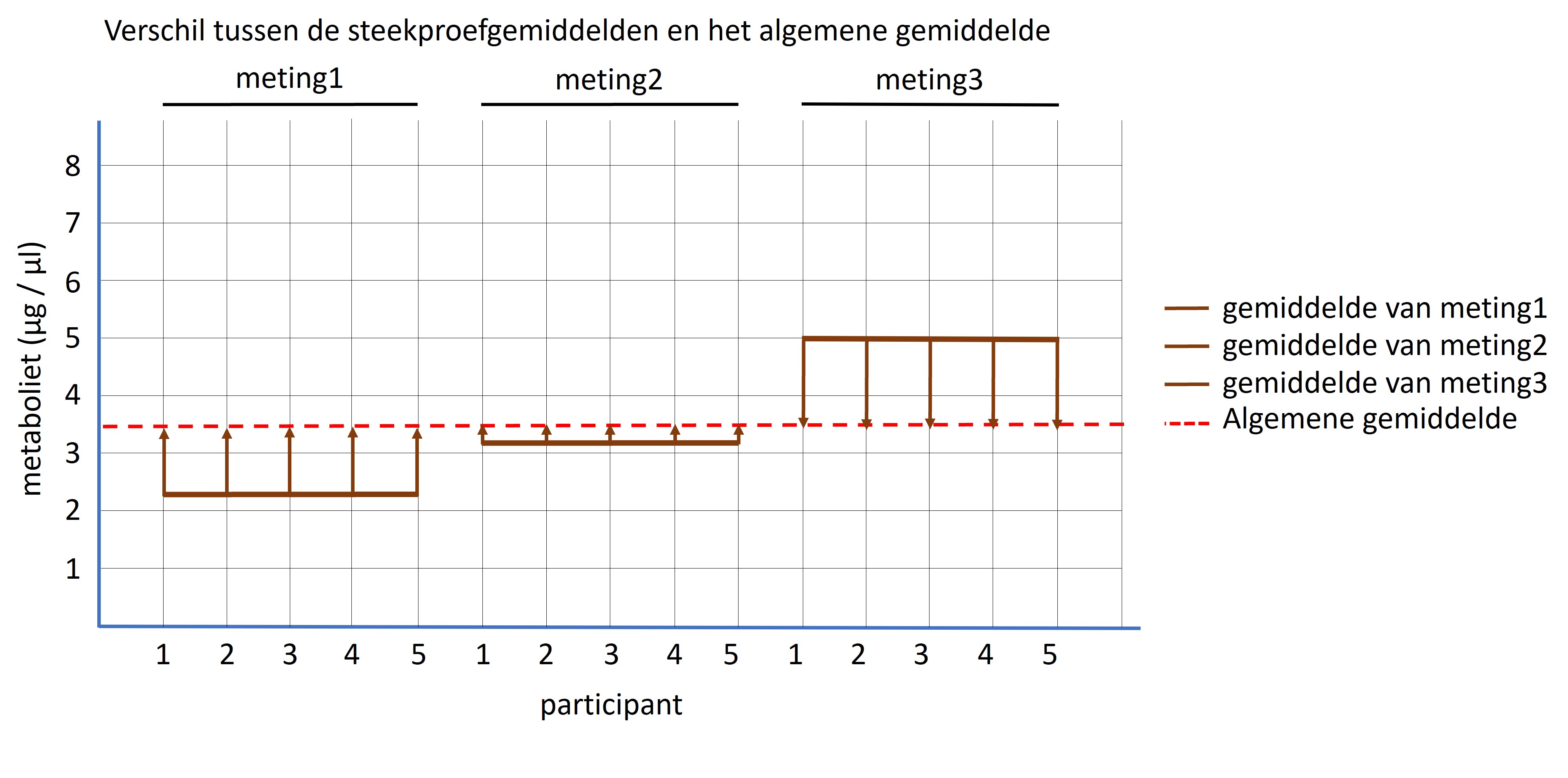
Figure 65: Verschil tussen het algemene gemiddelde en gemiddelde van iedere meting
Hoe meer het gemiddelde van meting1, meting2 en meting3 onderling van elkaar verschillen (de behandeling heeft een bepaald effect) hoe groter het verschil tussen het gemiddelde van iedere meting en het algemene gemiddelde. Dit verschil wordt uitgedrukt met de variantie waarbij het verschil wordt berekend tussen het gemiddelde van iedere meting en het algemene gemiddelde (zie figuur 65, aangegeven met de verticale bruine pijlen). Dit verschil wordt gekwadradeerd en vermenigvuldigd met de grootte van de steekproef (n) en van alle metingen bij elkaar opgeteld. We delen deze waarde ook door het aantal vrijheidsgraden, wat overeenkomt met (het aantal metingen -1) = 3 -1 = 2. Deze waarde is dus het signaal van het experiment en hoe groter het signaal, hoe groter de kans dat de experimentele behandeling een effect heeft gehad.
De tweede stap is het berekenen van de “variantie binnen de groepen” (noemer van de formule) en dit is de ruis van het experiment. De ruis wordt uitgedrukt door het verschil van ieder datapunt ten opzichte het gemiddelde van iedere meting uit te rekenen zoals bij een ongepaarde ANOVA. Echter omdat het een gepaarde meting is wordt de ruis hiervoor gecorrigeerd (dit wordt verder niet uitgelegd in deze cursus) waardoor de ruis kleiner wordt. Daarom is een gepaard experimenteel ontwerp gevoeliger om statistisch significante verschillen te detecteren omdat de ruis kleiner is en daardoor de signaal-ruis verhouding groter wordt in vergelijking met een ongepaarde ANOVA. De ruis wordt verder gedeeld door het aantal vrijheidsgraden.
Vervolgens berekenen we de F-statistiek zoals bij een ongepaarde ANOVA en wordt m.b.v. de F-verdeling uitgerekend wat de kans is om een bepaalde F-waarde te meten onder de aanname dat de behandeling niet heeft gewerkt. Als een behandeling niet werkt, is het signaal min of meer gelijk aan de ruis van het experiment en ligt de F-waarde rond de 1. Dit wordt formeel vastgelegd met de nul-hypothese (H0):
H0: \(\mu1\) = \(\mu2\) = \(\mu3\) (de gemiddelden van iedere meting zijn gelijk)
H1: De gemiddelden van de metingen zijn niet gelijk
(Deze hypothese stelt dat er ergens een verschil is tussen de groepen maar geeft niet aan tussen welke groepen!)
Intiutief voel je aan dat de kans steeds kleiner wordt om per toeval grote verschillen te meten tussen gepaarde metingen. Als deze kans (p-waarde) kleiner is dan 0,05 dan verwerpen we de H0 en wordt de H1 geaccepteerd. Er is wel een verschil tussen de gemiddelde van de metingen. Dit verschil berust niet op toeval.
P-waarde van de t-test ≥ 0,05: accepteer H0. Er is geen statistisch significant verschil tussen metingen
P-waarde van de t-test < 0,05: verwerp H0 en accepteer H1. Er is wel statistisch significant verschil tussen de metingen
We gebruiken het voorbeeld van hierboven om een gepaarde ANOVA uit te voeren. De aanwezigheid van een marker eiwit (voor chronische ontsteking) in het bloed van 6 personen is gemeten voor en na de verschillende tijdstippen van het trainingsschema.
Tabel I: Data van een gepaarde opzet. Ieder persoon is drie keer gemeten:
| persoon | meting1 | meting2 | meting3 |
|---|---|---|---|
| 1 | 45 | 50 | 55 |
| 2 | 42 | 42 | 45 |
| 3 | 36 | 41 | 43 |
| 4 | 39 | 35 | 40 |
| 5 | 51 | 55 | 59 |
| 6 | 44 | 49 | 56 |
Het stappenplan voor het uitvoeren van een gepaarde ANOVA is hetzelfde als voor een ongepaarde ANOVA:
- Laad de libraries: die zijn al geladen van de vorige voorbeelden en opdrachten
(2a) Laad de data in R en maak de data tidy
(2b) Zet de kolom met de onafhankelijke variabele (=factor) om naar een factor variabele
# Laad de data in als een tibble
protein <- tibble(persoon=c(1,2,3,4,5,6),
meting1=c(45, 42, 36, 39, 51, 44),
meting2=c(50, 42, 41, 35, 55, 49),
meting3=c(55, 45, 43, 40, 59, 56))
# Maak de data tidy
protein_tidy<-protein |>
pivot_longer(cols=c("meting1","meting2","meting3"),
names_to="meting",
values_to="concentration")
# Zet de kolommen met de onafhankelijke variabele (=factor) om naar een factor variabele
protein_tidy$persoon <-as.factor(protein_tidy$persoon)
protein_tidy$meting <-as.factor(protein_tidy$meting)- Beschrijvende statistiek
# Bereken gemiddelde, sd en het aantal metingen per groep
protein_tidy_summarize <-protein_tidy |>
group_by(meting) |>
summarize(mean_concentration=mean(concentration),
sd_concentration=sd(concentration),
n_concentration=length(concentration))
# Aantal metingen is 6 per groep. (4a) Test of de data normaal is verdeeld
Hiervoor gebruiken we niet de ruwe data (meetpunten per experimentele groep) maar de residuals (zie figuur 63, maar dan voor iedere gepaarde meting in plaats van iedere onafhankelijke groep).
De eerste stap is het verkrijgen van de residuals:
# Verkrijg de residuals met behulp van de aov() functie
model_protein <- aov(data = protein_tidy,
concentration ~ meting + Error(persoon/meting) ) # De error term in het model berekent de residuals
res_protein <- model_protein$`persoon:meting`$residuals # Extract residuals van het model_protein object
# maak een tibble aan met de residuals
res_protein_tibble <- tibble(res=res_protein)(4b) Test of de residuals normaal verdeeld zijn met Shapiro-Wilk test en qq-plot
# Test of de residuals normaal verdeeld zijn met de Shapiro-Wilk test
shapiro.test(res_protein_tibble$res)$p.value |> round(digits=2)
# Maak een qq-plot met de residuals
res_protein_tibble |>
ggplot(aes(sample = res)) +
stat_qq() +
stat_qq_line(color = "red") +
theme_minimal()
# Conclusie. P-waarde SW test is 0.26. Accepteer H~0~. De data is normaal verdeeld
# Dit wordt verder ondersteund met de qq-plot(4c) Optioneeel (want data is al normaal verdeeld): Test of de data uitbijters bevat:
protein_tidy |>
ggplot(aes(x=meting, y=concentration))+
geom_boxplot()
# conclusie: geen uitbijters in de boxplot- Visualiseer de data
# Staafdiagram + errorbars met behulp van ggplot
protein_tidy_summarize |> ggplot( aes(x = meting, y = mean_concentration)) +
geom_col() +
geom_errorbar(
aes(ymin = mean_concentration - sd_concentration,
ymax = mean_concentration + sd_concentration),
width = 0.2) +
labs(
x = "meting",
y = "concentration (pg/ml)",
title = "Effect van oefeningen op chronische stress") +
theme_minimal()(6 + 7) Test of de varianties tussen de groepen gelijk zijn en voer een gepaarde ANOVA test uit
(Dit wordt tegelijkertijd uitgevoerd met de aov_ex() functie van de afex package)
Om een gepaarde ANOVA uit te voeren gebruiken we het “Analysis of Factorial Experiments” (afex) packages:
if (!require("afex")) {
install.packages("afex")
library(afex)
}De aov_ez() functie van het afex package voert standaard de “Mauchly Tests” uit om te testen of de varianties gelijk zijn. Als de p-waarde van deze test kleiner is dan 0,05 dan zijn de varianties niet gelijk. Als de varianties niet gelijk zijn wordt er een Greenhouse–Geisser (GG) of een Huynh–Feldt (HF) correctie toegepast op de p-waarde van de gepaarde ANOVA. Na de correctie wordt de p-waarde van de gepaarde ANOVA test groter en dus conservatiever. Het is aan te raden om altijd de gecorrigeerde waarde af te lezen van de gepaarde ANOVA, zeker bij kleine steekproeven die minder dan 20 metingen bevat.
gepaarde_anova <- aov_ez(
data = protein_tidy,
id = "persoon",
dv = "concentration",
within = "meting")
summary(gepaarde_anova)##
## Univariate Type III Repeated-Measures ANOVA Assuming Sphericity
##
## Sum Sq num Df Error SS den Df F value Pr(>F)
## (Intercept) 37996 1 658.28 5 288.602 1.293e-05 ***
## meting 143 2 57.22 10 12.534 0.001886 **
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
##
## Mauchly Tests for Sphericity
##
## Test statistic p-value
## meting 0.43353 0.18795
##
##
## Greenhouse-Geisser and Huynh-Feldt Corrections
## for Departure from Sphericity
##
## GG eps Pr(>F[GG])
## meting 0.63838 0.008985 **
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## HF eps Pr(>F[HF])
## meting 0.760165 0.005284458In dit voorbeeld is de p-waarde van de “Mauchly Tests for Sphericity” 0.18795. Zoals hierboven beschreven is de Mauchly Tests onnauwkeurig bij minder dan 20 metingen en in dit voorbeeld zijn er 6 metingen. Dus hoewel we aan de hand van de p-waarde kunnen concluderen dat de varianties gelijk zijn (want p-waarde > 0,05) en we dus de normale p-waarde (0.001886) kunnen aflezen is dit niet correct. We lezen de Greenhouse–Geisser (GG, p-waarde=0.008985) of de Huynh–Feldt (HF, p-waarde=0.005284458) p-waarde af zoals hierboven weergegeven:
Beide p-waarden zijn < 0.05 dus de H0 hypothese wordt verworpen en de de H1 hypothese wordt geaccepteerd. Er is wel een verschil tussen de drie metingen. Maar we weten niet tussen welke metingen er een statistisch significant verschil is. Hiervoor gebruiken we weer een post-hoc test om alle combinaties te testen. We gebruiken hiervoor het package “emmeans”
Post-hoc test: gepaard
emmeans(gepaarde_anova,
pairwise ~ meting,
adjust = "bonferroni")## $emmeans
## meting emmean SE df lower.CL upper.CL
## meting1 42.8 2.12 5 37.4 48.3
## meting2 45.3 2.97 5 37.7 53.0
## meting3 49.7 3.24 5 41.3 58.0
##
## Confidence level used: 0.95
##
## $contrasts
## contrast estimate SE df t.ratio p.value
## meting1 - meting2 -2.50 1.520 5 -1.643 0.4842
## meting1 - meting3 -6.83 1.700 5 -4.017 0.0305
## meting2 - meting3 -4.33 0.715 5 -6.061 0.0053
##
## P value adjustment: bonferroni method for 3 testsIn de output zien we onder “contrasts” een statistisch significant verschil tussen:
meting1 - meting3 (p-waarde = 0.0305)
meting2 - meting3 (p-waarde = 0.0053)
les8_opdracht
Een student wil een protocol optimaliseren voor eiwitproductie. De student induceert bacteriëen zodat een specifiek eiwit aangemaakt wordt. Om het eiwit te zuiveren vanuit het bacteriëel cellysaat moet het lysaat op een zuiveringskolom worden gebracht zodat alleen het specifieke eiwit achterblijft op de kolom en alle andere eiwitten eraf worden gewassen. De student wil drie verschillende kolommen testen om te bepalen wat de hoogste opbrengst geeft. Het bacteriëel celllysaat wordt verdeeld in drie groepen en getest met de verschillende kolommen. De eiwit opbrengst (mg / L) data staat in bestand les8_opdracht3 in de gedeelde map op de server.
Is er een verschil in opbrengst tussen de 3 kolommen?
Klik voor het antwoord
# Het is een gepaarde meting want hetzelfde monsters wordt gesplitst...
# en drie keer gemeten (op verschillende kolommen)
# (1) Laad de libraries
library(tidyverse)
library(ggplot2)
library(moments)
library(afex)
library(emmeans)
# (2a) Laad de data in R. Let op: de kolommen zijn gescheiden door een semicolon (;)
# Gebruik read_csv2() van tidyverse om de data in te lezen
kolom <- read_csv2("data/les8/les8_opdracht3.txt",
locale=locale(decimal_mark = ","))
# (2b) maak de data tidy
kolom_tidy <- kolom|>
pivot_longer(cols=c("kolom1","kolom2","kolom3"),
names_to="kolom",
values_to="yield")
# (2c) Zet de kolom met de onafhankelijke variabele (=factor) om naar een factor variabele
kolom_tidy$lysaat <- as.factor(kolom_tidy$lysaat)
kolom_tidy$kolom <- as.factor(kolom_tidy$kolom)
# (3) beschrijvende statistiek: gemiddelde, sd en aantal metingen
kolom_tidy_summarize <-kolom_tidy |>
group_by(kolom) |>
summarize(mean_yield=mean(yield),
sd_yield=sd(yield),
n_yield=length(yield))
# (4a) Verkrijg de residuals met behulp van de aov() functie
model_kolom <- aov(data = kolom_tidy,
yield ~ kolom + Error(lysaat/kolom) ) # De error term in het model berekent de residuals
res_kolom <- model_kolom$`lysaat:kolom`$residuals # Extract residuals van het model_kolom object
# maak een tibble aan met de residuals
res_kolom_tibble <- tibble(res=res_kolom)
# (4b) # Test of de residuals normaal verdeeld zijn met de Shapiro-Wilk test
shapiro.test(res_kolom_tibble$res)$p.value |> round(digits=2)
# Maak een qq-plot met de residuals
res_kolom_tibble |>
ggplot(aes(sample = res)) +
stat_qq() +
stat_qq_line(color = "red") +
theme_minimal()
# Conclusie. P-waarde SW test is 0.73. Accepteer H~0~. De data is normaal verdeeld
# Dit wordt verder ondersteund met de qq-plot. De meeste punten op de referentie lijn
# (4c) Optioneeel (want data is al normaal verdeeld): Test of de data uitbijters bevat:
kolom_tidy |>
ggplot(aes(x=kolom, y=yield))+
geom_boxplot()
# Conclusie: geen uitbijters
# (5) Visualiseer de data
# Staafdiagram + errorbars met behulp van ggplot
kolom_tidy_summarize |> ggplot( aes(x = kolom, y = mean_yield)) +
geom_col() +
geom_errorbar(
aes(ymin = mean_yield - sd_yield,
ymax = mean_yield + sd_yield),
width = 0.2) +
labs(
x = "kolom type",
y = "yield (mg / l)",
title = "Effect van zuiveringkolom op de opbrengst") +
theme_minimal()
# (6+7) Testen of varianties gelijk zijn en uitvoeren van de gepaarde anova
# Er zijn maar 5 monsters gemeten dus gebruik de GG of HF gecorrigeerde p-waarde
gepaarde_anova <- aov_ez(
data = kolom_tidy,
id = "lysaat", # welke monster er gebruikt is
dv = "yield", # afhankelijke variabele
within = "kolom") # onafhankelijke variabele (in dit geval de verschillende kolommen)
summary(gepaarde_anova)
# Conclusie: de GG (0.6298) en HF (0.6615738) gecorrigeerde p-waarden > 0.05. Accepteer H~0~.
# Er is geen statistisch significant verschil in de gemiddelde yield tussen de drie kolommenTwo-way ANOVA
Gebruik de “two-way ANOVA” bij een experiment met “multiple factors” zie les4.
In les4_opdracht2 hebben we het volgende voorbeeld gebruikt:
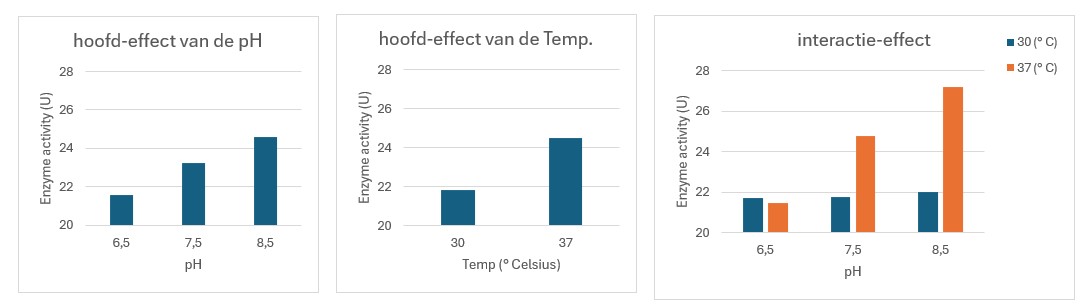
Figure 66: Visualisatie van hoofd- en interactie-effecten
Met een two-way ANOVA testen we drie verschillende null hypothesen:
Factor A: H0: \(\mu1\) = \(\mu2\) met de alternatieve hypothese H1: \(\mu1\) ≠ \(\mu2\)
Factor B: H0: \(\mu1\) = \(\mu2\) met de alternatieve hypothese H1: \(\mu1\) ≠ \(\mu2\)
Interactie H0: Het effect van factor A is gelijk voor alle ‘levels’ van factor B
Interactie H1: Het effect van factor A is niet gelijk voor alle ‘levels’ van factor B
OPMERKING: Deze null hypotheses zijn voor twee factoren met ieder twee levels. Als een factor bijvoorbeeld 3 levels heeft dan worden de hypothesen \(\mu1\) = \(\mu2\) = \(\mu3\) en voor de H1 de gemiddelden van de groepen zijn niet gelijk (maar we weten niet tussen welke groepen = (levels)).
De two-way ANOVA wordt uitgevoerd met de aov() functie. We demonstreren de two-way ANOVA aan de hand van de data van figuur 66. De data van deze figuur staat in bestand les8_enzym_activiteit.csv in de gedeelde folder.
- Laad de libraries: die zijn al geladen van de vorige voorbeelden en opdrachten
(2a) Laad de data in R, indien nodig maak de data tidy
(2b) Zet de kolom met de onafhankelijke variabele (=factor) om naar een factor variabele
# Inladen van de data. De data is een tsv bestand.
# Het decimaalteken is een punt. De data is al tidy
enzym_tidy <- read_tsv("data/les8/les8_enzym_activiteit.csv")
# Zet de kolommen met de onafhankelijke variabele (=factor) om naar een factor variabele
enzym_tidy$meting <-as.factor(enzym_tidy$meting)
enzym_tidy$temp <-as.factor(enzym_tidy$temp)
enzym_tidy$ph <-as.factor(enzym_tidy$ph)- Beschrijvende statistiek
# Bereken gemiddelde, sd en het aantal metingen per groep
enzym_tidy_summarize <-enzym_tidy |>
group_by(temp, ph) |>
summarize(mean_activiteit=mean(activiteit),
sd_activiteit=sd(activiteit),
n_activiteit=length(activiteit))
# Aantal metingen is 5 per groep. (4a) Test of de data normaal is verdeeld
Hiervoor gebruiken we niet de ruwe data (meetpunten per experimentele groep) maar de residuals (zie figuur 63). De eerste stap is het verkrijgen van de residuals:
# Verkrijg de residuals met behulp van de aov() functie
model_enzym <- aov(data = enzym_tidy,
activiteit ~ temp * ph)
res_enzym <- residuals(model_enzym) # Extract residuals van het model_enzym object
# maak een tibble aan met de residuals
res_enzym_tibble <- tibble(res=res_enzym)(4b) Test of de residuals normaal verdeeld zijn met Shapiro-Wilk test en qq-plot
# Test of de residuals normaal verdeeld zijn met de Shapiro-Wilk test
shapiro.test(res_enzym_tibble$res)$p.value |> round(digits=2)
# Maak een qq-plot met de residuals
res_enzym_tibble |>
ggplot(aes(sample = res)) +
stat_qq() +
stat_qq_line(color = "red") +
theme_minimal()
# Conclusie. P-waarde SW test is 0.35. Accepteer H~0~. De data is normaal verdeeld
# Dit wordt verder ondersteund met de qq-plot(4c) Optioneeel (want data is al normaal verdeeld): Test of de data uitbijters bevat:
enzym_tidy |> ggplot(aes(x = ph, y = activiteit, fill = temp)) +
geom_boxplot() +
labs(title="data verdeling van de 6 experimentele groepen",
x = "Temperatuur (°C)",
y = "Activiteit",
fill = "pH") +
theme_minimal()
# conclusie: 1 uitbijter in de boxplot van combinatie pH 7.5 en Temp = 30 °C
# Data is normaal verdeel dus de uitbijter wordt niet verwijderd- Visualiseer de data
enzym_tidy_summarize |> ggplot(aes(x = ph, y = mean_activiteit, fill = temp)) +
geom_col(position = position_dodge(width = 0.8)) +
geom_errorbar(aes(ymin = mean_activiteit - sd_activiteit,
ymax = mean_activiteit + sd_activiteit),
width = 0.2,
position = position_dodge(width = 0.8)) +
labs(x = "pH", y = "Activiteit", fill = "Temperatuur") +
theme_minimal()- Test of de varianties tussen de groepen gelijk zijn met de Levene’s test
# test of de variantie gelijk zijn met de Levene's test
leveneTest(data = enzym_tidy,
activiteit ~ temp * ph)
# conclusie: p-waarde = 0.6386. Accepteer H~0~. Varianties zijn gelijk- voer een two-way ANOVA test uit
# Uitvoeren van de two-way ANOVA
model_enzym <- aov(data = enzym_tidy, # data bestand
activiteit ~ temp * ph) # formule die de afhankelijke variabele (links) en de twee onafhankelijke variabelen (rechts) aangeeft
summary(model_enzym)## Df Sum Sq Mean Sq F value Pr(>F)
## temp 1 52.54 52.54 30.29 1.17e-05 ***
## ph 2 45.48 22.74 13.11 0.000142 ***
## temp:ph 2 37.73 18.87 10.88 0.000434 ***
## Residuals 24 41.62 1.73
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1De output noteert voor iedere H0 een p-waarde:
Factor A (Temp): p-waarde = 1.17e-05
Factor B (pH): p-waarde = 0.000142
Interactie tussen A en B (Temp:pH) : p-waarde = 0.000434
De p-waarden zijn allemaal < 0,05: verwerp H0 en accepteer H1:
Factor A (Temp) heeft een statistisch significant effect op de enzym activiteit.
Factor B (pH) heeft een statistisch significant effect op de enzym activiteit.
Er is een interactie tussen factor A (Temp) en factor B (pH): de pH heeft alleen maar een effect bij 37 °C en niet bij 30 °C. De temperatuur beinvloed het effect van de pH.
Non-parametrische toetsen
Een niet parametrische toets (zie figuur 59, rechterkant van de beslisboom en de tabel hieronder) wordt gebruikt als:
(1) De data van minimaal één (of meerdere) experimentele groepen niet normaal verdeeld is
(2) Het type data nominaal of ordinaal is
Deze toetsen zijn gebaseerd op de mediaan in plaats van het gemiddelde. Non-parametrische toetsen zijn minder gevoelig om statistisch significante verschillen aan te tonen. Omdat in non-parametrische testen niet het gemiddelde wordt gebruikt, maar de mediaan wordt de nul hypothese anders geformuleerd:
H0: De verdeling van de experimentele groepen zijn gelijk verdeeld
H1: De verdeling van de experimentele groepen zijn niet gelijk verdeeld
Anders geformuleerd, er is geen (H0) of wel (H1) verschil tussen experimentele groepen. De interpretatie van de hypotheses zijn dus gelijk van parametrische en non-parametrische toetsen.
| Parametrische toest | Non-parametrische variant |
|---|---|
| Two samples unpaired T-test | Mann-Whitney |
| Two samples paired T-test | Wilcoxin SR |
| Unpaired one Way ANOVA | Kruskal Wallis |
| Paired one Way ANOVA | Friedman |
Mann-Whitney
Gebruik de “Mann-Whitney test” bij een ongepaard OFAT experiment waarbij er twee onafhankelijke groepen zijn die ieder één keer zijn gemeten en waarvan de data niet normaal verdeeld is of het type data is nominaal of ordinaal.
De Mann-Whitney test wordt uitgevoerd met de wilcox.test() functie. We demonstreren aan de hand van de data die aanwezig is in R-object les8_mann_whitney.RDS. R-objecten (met de extensie .RDS) kunnen ingelezen worden met de readRDS() functie (zie ook ibip cursus les1: Data inlezen. Het R-object staat in de gedeelde folder.
Dit object bevat data van een scratch assay. De data is niet normaal verdeeld
# inlezen van een R-object. De data is tidy
scratch_mann_whitney <- readRDS("data/les8/les8_mann_whitney.RDS")
# data inspectie: welke kolommen zijn aanwezig
names(scratch_mann_whitney)
# de kolom 'groep' omzetten naar een factor variabele (indien nodig)
scratch_mann_whitney$groep <- as.factor(scratch_mann_whitney$groep)# Mann-whitney test op tidy data
wilcox.test(data=scratch_mann_whitney,
area_perc ~ groep)##
## Wilcoxon rank sum test with continuity correction
##
## data: area_perc by groep
## W = 205, p-value = 0.0001124
## alternative hypothesis: true location shift is not equal to 0# conclusie: p-waarde = 0.0001124. Verwerp de H~0~ en accepteer de H~1~.
# De verdelingen zijn niet gelijk. Er is wel een verschil tussen de experimentele groepenWilcoxon SR test
Gebruik de “Wilcoxon SR” bij een gepaard OFAT experiment waarbij waarbij één groep twee keer wordt gemeten en waarvan de data niet normaal verdeeld is of het type data is nominaal of ordinaal.
De Wilcoxon SR wordt uitgevoerd met de wilcox.test() functie. We demonstreren aan de hand van de data die aanwezig is in R-object les8_wilcoxonsr.RDS. Het R-object staat in de gedeelde folder.
De data is afkomstig van patiënten waarbij de tumorgrootte is gemeten voor en na de behandeling met een cytotoxische stof.
De data is niet normaal verdeeld.
# inlezen van een R-object. De data is niet tidy
tumor_wilcoxon <- readRDS("data/les8/les8_wilcoxonrs.RDS")
# data inspectie: welke kolommen zijn aanwezig
names(tumor_wilcoxon)
wilcox.test(tumor_wilcoxon$voor,
tumor_wilcoxon$na,
paired=TRUE)## Warning in wilcox.test.default(tumor_wilcoxon$voor, tumor_wilcoxon$na, paired
## = TRUE): cannot compute exact p-value with ties# conclusie: p-waarde = 0.0002962. Verwerp de H~0~ en accepteer de H~1~.
# De verdelingen zijn niet gelijk. Er is wel een verschil tussen de experimentele groepenKruskall Wallis
Gebruik de “Kruskall Wallis test” bij een ongepaard OFAT experiment waarbij er drie of meer onafhankelijke groepen zijn, die ieder één keer zijn gemeten en waarvan de data niet normaal verdeeld is of het type data is nominaal of ordinaal.
De Kruskall Wallis wordt uitgevoerd met de kruskal.test() functie. We demonstreren aan de hand van de data die aanwezig is in R-object les8_kruskal.RDS. Het R-object staat in de gedeelde folder.
De data is afkomstig van een experiment waarbij de enzymactiviteit is gemeten in drie verschillende cell culturen die niet gestimuleerd zijn (controle) of gestimuleerd zijn met drug A of drug B.
De data is niet normaal verdeeld.
# inlezen van een R-object. De data is tidy
enzym_activiteit <- readRDS("data/les8/les8_kruskal.RDS")
# data inspectie: welke kolommen zijn aanwezig
names(enzym_activiteit)
# de kolom 'treatment' omzetten naar een factor variabele (indien nodig)
enzym_activiteit$treatment <- as.factor(enzym_activiteit$treatment)# kruskal-wallis test op tidy data
kruskal.test(data=enzym_activiteit,
activity ~ treatment)##
## Kruskal-Wallis rank sum test
##
## data: activity by treatment
## Kruskal-Wallis chi-squared = 12.394, df = 2, p-value = 0.002036# conclusie: p-waarde = 0.002036. Verwerp de H~0~. Accepteer de H~1~
# De verdelingen zijn niet gelijk. Er is een verschil tussen de experimentele groepen.
# Maar we weten niet tussen welke groepen er een verschil is -> post-hoc testVoor een Kruskal-Wallis post-hoc test wordt de functie pairwise.wilcox.test() gebruikt in combinatie met de ‘paired=FALSE’ parameter. De correctie methode is Benjamini-Hochberg
pairwise.wilcox.test(enzym_activiteit$activity,
enzym_activiteit$treatment,
paired=FALSE, # geeft aan dat het een ongepaarde meting is
p.adjust.method = "BH") # de correctie methode is Benjamini-Hochberg##
## Pairwise comparisons using Wilcoxon rank sum exact test
##
## data: enzym_activiteit$activity and enzym_activiteit$treatment
##
## Control DrugA
## DrugA 0.7788 -
## DrugB 0.0036 0.0036
##
## P value adjustment method: BH# Conclusie: Er is een statistisch significant verschil tussen experimentele groepen:
# controle - drugB (p-waarde = 0.0036)
# drugA - drug B (p-waarde = 0.0036)Friedman
Gebruik de “Friedman test” bij een gepaard OFAT experiment waarbij één groep meerdere keren wordt gemeten en waarvan de data niet normaal verdeeld is of het type data is nominaal of ordinaal.
De Friedman test wordt uitgevoerd met de friedman.test functie. We demonstreren aan de hand van de data die aanwezig is in R-object les8_friedman.RDS. Het R-object staat in de gedeelde folder.
De data is afkomstig van een experiment waarbij de neurale activiteit herhaaldelijk is gemeten in ratten met alzheimer die een bepaald parkoers lopen.
De data is niet normaal verdeeld.
# inlezen van een R-object. De data is tidy
alzheimer <- readRDS("data/les8/les8_friedman.RDS")
# data inspectie: welke kolommen zijn aanwezig
names(alzheimer)
# de kolom 'meting' omzetten naar een factor variabele (indien nodig)
alzheimer$meting <- as.factor(alzheimer$meting)# friedman test op tidy data
friedman.test(data=alzheimer,
activity ~ meting | rat) # Voor een gepaarde meting moet je ook wat herhaaldelijk is gemeten. In dit geval de rat##
## Friedman rank sum test
##
## data: activity and meting and rat
## Friedman chi-squared = 13.52, df = 2, p-value = 0.001159# conclusie: p-waarde = 0.001159. Verwerp de H~0~ . Accepteer H~1~
# De verdelingen zijn niet gelijk. Er is wel een verschil tussen de experimentele groepen.
# Maar we weten niet tussen welke groepen er een verschil is -> post-hoc testVoor een Friedman post-hoc test wordt de functie pairwise.wilcox.test() gebruikt in combinatie met de ‘paired=TRUE’ parameter. De correctie methode is Benjamini-Hochberg
pairwise.wilcox.test(alzheimer$activity,
alzheimer$meting,
paired=TRUE, # geeft aan dat het een gepaarde meting is
p.adjust.method = "BH") # de correctie methode is Benjamini-Hochberg##
## Pairwise comparisons using Wilcoxon signed rank exact test
##
## data: alzheimer$activity and alzheimer$meting
##
## meting1 meting2
## meting2 0.00036 -
## meting3 0.00137 0.38098
##
## P value adjustment method: BH# Conclusie: Er is een statistisch significant verschil tussen de metingen:
# meting1 - meting2 (p-waarde = 0.00036)
# meting2 - meting3 (p-waarde = 0.00137)Casus
De interne R-dataset “ToothGrowth” bevat gegevens uit een onderzoek naar het effect van vitamine C op de tandgroei (len) bij cavia’s. Het experiment is uitgevoerd met 60 cavia’s, waarbij elk dier één van de drie doseringen vitamine C (0,5, 1 en 2 mg/dag) kreeg toegediend via één van de twee toedieningsmethoden: sinaasappelsap (orange juice (OJ) of ascorbinezuur (VC).
Factor A: vitamine C concentratie met de levels 0.5, 1 en 2 mg / dag Factor B: Toedieningsmethode met de levels OJ (orange juice) en ascorbinezuur (VC) Afhankelijke variabele: tandgroei (len)
Bedenk eerst wat voor type experimentele proefopzet dit is. Test de verschillende nul hypothesis voor deze proefopzet